Avicenna Journal of Medical Biochemistry. 8(2):83-88.
doi: 10.34172/ajmb.2020.12
Research Article
Molecular Docking and Fragment-Based QSAR Modeling for In Silico Screening of Approved Drugs and Candidate Compounds Against COVID-19
Saeid Afshar 1  , Asrin Bahmani 1, Massoud Saidijam 1, *
, Asrin Bahmani 1, Massoud Saidijam 1, *
Author information:
1Research Center for Molecular Medicine, Hamadan University of Medical Sciences, Hamadan, Iran.
*
Corresponding author: Massoud Saidijam, Department of Molecular Medicine and Genetics, Research Center for Molecular Medicine, Hamadan University of Medical Sciences, Hamadan, Iran, Email:
sjam110@yahoo.com
Abstract
Background: Coronavirus disease 2019 (COVID-19) as a serious global health crisis leads to high mortality and morbidity. However, currently, there are no effective vaccines and treatments for COVID-19. Main protease (Mpro) and angiotensin-converting enzyme 2 (ACE2) are the best therapeutic targets of COVID-19.
Objectives: The main purpose of this study is to investigate the most appropriate drug and candidate compound for proper interaction with Mpro and ACE2 to inhibit the activity of COVID-19.
Methods: In this study, repurposing of approved drugs and screening of candidate compounds using molecular docking and fragment-based QSAR method were performed to discover the potential inhibitors of Mpro and ACE2. QSAR and docking calculations were performed based on the prediction of the inhibitory activities of 5-hydroxy indanone derivatives. Based on the results, an optimal structure was proposed to inhibit the activity of COVID-19.
Results: Among 2629 DrugBank approved drugs, 118 were selected considering the LibDock score and absolute energy for possible drug-Mpro interactions. Furthermore, the top 40 drugs were selected based on screening the results for possible drug- Mpro interactions with AutoDock Vina.
Conclusion: Finally, evaluation of the top 40 selected drugs for possible drug-ACE2 interactions with AutoDock Vina indicated that deslanoside (DB01078) can interact effectively with both Mpro and ACE2. However, prior to conducting clinical trials, further experimental validation is needed.
Keywords: COVID-19, Main Protease, ACE2, Drug repurposing, Fragment-QSAR
Copyright and License Information
© 2020 The Author(s); Published by Hamadan University of Medical Sciences.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium provided the original work is properly cited.
Background
In December 2019, a novel coronavirus entitled SARS-coV2 emerged in Wuhan, China, led to a rapid spread of pulmonary disease named coronavirus disease 2019 (COVID-19) (1). COVID-19 pandemic is a serious global health crisis. As of 16 November, 2020, more than 54.3 million confirmed cases of COVID-19 and more than 1.316 million confirmed deaths due to this disease were reported globally, according to the World Health Organization (WHO). However, in spite of extensive research being conducted by several research groups in the field of vaccine and drug design, there are currently no certain treatments for COVID-19 (2).
SARS-CoV2 as a member of Betacoronaviruses possesses a relatively large single-strand RNA (3). The viral genome comprises structural and non-structural genes. The structural genes encode four proteins including the spike (S), nucleocapsid (N), envelope (E), and membrane (M) proteins. The main protein products of nonstructural genes are the main protease (Mpro ), RNA dependent RNA polymerase (RdRP), and papain-like protease (PLpro) (4,5).
Among different therapeutic targets of SARS-COV-2, Mpro and angiotensin-converting enzyme 2 (ACE2) are the best-studied targets (1,6). Proteolytic activity of Mproenzymehas an essential role in viral replication due to the viral polyprotein processing (7). On the other hand, the attachment of S protein to ACE2 is critical for the recognition and entrance of the virus into the host cell (8). Therefore, the inhibition of Mproactivity leads to the prevention of viral replication and preventing the attachment of S protein to ACE2 of the host cell prevents the entrance of virus to the host cell (7,8).
In recent years, the use of bioinformatics for drug design has been considered by researchers. The application of bioinformatics for predicting, analyzing, or interpreting clinical and para-clinical findings is of great importance (9).
Marinho et al conducted a virtual screening study to investigate the molecular docking of possible inhibitors of COVID-19 main protease. The results of their study showed that all inhibitors bind to the same enzyme site, specifically in domain III of the SARS-CoV-2 main protease. Therefore, they suggested baricitinib and quinacrine, in combination with azithromycin (10).
Vardhan and Sahoo used in silico ADMET and molecular docking to study the inhibitory activity of limonoids and triterpenoids against COVID-19. The study of protein-ligand interactions showed that these phytochemicals bind to amino acid residues at the active site of target proteins. Therefore, the core structure of these potential hits can be used for further lead optimization to design drugs for SARS-CoV-2 (11).
Abo-Zeid et al conducted a docking study of the interaction of IONPs (Fe2O3 and Fe3O4) with the binding domain of the spike protein receptor (S1-RBD) of SARS-CoV-2. Their studies revealed that both Fe2O3 and Fe3O4 interacted efficiently with the SARS-CoV-2 S1-RBD and HCV glycoproteins, E1 and E2. Fe3O4 formed a more stable complex with S1-RBD whereas Fe2O3 favored HCV E1 and E2 (12).
By considering the essential role of these therapeutic targets (Mpro and ACE2), repurposing of approved drugs and screening of candidate compounds using modeling methods such as molecular docking and fragment-based QSAR method were performed to discover the potential inhibitors of Mpro and ACE2.
Materials and Methods
Computer Hardware and Software
The molecular structures of all compounds were drawn with the HyperChem 8.0 (Hypercube, Inc., Gainesville, 2011) and pre-optimized using the MM+ molecular mechanics (Polak–Ribiere algorithm). The final geometries of the minimum energy conformation were obtained by more precise optimization with the semi-empirical PM3 method, applying a root mean square gradient limit of 0.05 (kcal.mol-1 Å-1) as a stopping criterion for optimized structures. The molecular fragments descriptors were calculated by ISIDA/QSPR software (version 5.88.012, 2015). A three-step technique was used for the selection of descriptors using ISIDA and other calculations were performed using Molegro Data Modeler (MDM, 2009, 2.1.0). The Discovery Studio (Version 2016) was used for the evaluation of LibDock score. AutoDock Tools 1.5.6 was used for preparation of ligand and receptor for docking and AutoDock Vina (version 1.1.2) was used for binding free energy calculation.
Ligand Screening for ACE2 and Mpro
In the first stage, 2629 approved drugs were retrieved from the DrugBank database (version 5.1.5,https://www.drugbank.ca/). The targets used for further in silico screening were 3D structure of COVID-19 Mpro (PDB ID: 6W63) and 3D structure of ACE2 receptor (PDB ID: 6LZG) which were retrieved from the PDB database (https://www.rcsb.org/). The 3D molecular structures of all approved drugs were generated and their energy was minimized using Discovery Studio (version 2016). The initial virtual screening of compounds for possible drug-Mpro interactions was done by the LibDock module of Discovery Studio (version 2016). Then, the top 118 approved drugs were selected for further evaluation with AutoDock Vina (version 1.1.2). Afterwards, the top 40 selected drugs were evaluated for possible drug-ACE2 interactions with AutoDock Vina (Ver.1.1.2).
Evaluation of Derivatives of 5-Hydroxy Indanone
In 2018, Vulupala et al designed and synthesized 5-hydroxy indanone derivatives under click chemistry reaction conditions. Synthesized derivatives were confirmed by biological evaluation as ACE inhibitors. The results of their study showed that 5-hydroxy indanone derivatives with minimal toxicity were comparable to the clinical drug lisinopril (13). Therefore, we decided to use 5-hydroxy indanone derivatives as base compounds in QSAR calculations as these compounds have the potential to inhibit the activity of ACE2. In the second stage of the calculations of this study, the derivatives of 5-hydroxy indanone were used as the basis for the fragment-based QSAR and molecular docking calculations. The calculation steps were as follows: First, the QSAR calculations were performed using ISIDA/QSPR software. Then, the percentages of inhibition of the compounds were predicted, and the most effective fragments were identified. In the next step, the selected compounds were included in the molecular docking calculations and were evaluated according to the conditions of the approved drugs. Finally, one or more compounds were introduced as compounds that have the potential to inhibit Mproand ACE2 activity.
Results
Ligand Screening for ACE2 and Mpro
After the first virtual screening, 118 approved drugs were selected by considering the LibDock score and absolute energy. Then, the top 40 compounds were selected based on the screening results for possible drug-Mpro interactions with AutoDock Vina. Further evaluation of the final selected compounds for possible drug-ACE2 interactions with AutoDock Vina indicated that deslanoside (DB01078) can interact effectively with both Mproand ACE2. Table 1 shows the LibDock scores and binding energies of the top 10 selected compounds for Mproand ACE2.
Table 1.
LibDock Scores and Binding Energies of the Top 10 Selected Compounds for Mproand ACE2
|
Drug Name
|
DrugBank
ID
|
LibDock score
|
The Binding Energy for M
pro
(kcal/mol)
|
The Binding Energy for ACE2 (kcal/mol)
|
| Ergotamine |
DB00696 |
153.418 |
-9.4 |
-7.7 |
| Deslanoside |
DB01078 |
184.133 |
-9.3
|
-9
|
| Plicamycin |
DB06810 |
199.643 |
-9.1 |
-6.3 |
| Glecaprevir |
DB13879 |
155.201 |
-9.1 |
-7.1 |
| Irinotecan |
DB00762 |
138.15 |
-9.1 |
-8.2 |
| Voxilaprevir |
DB12026 |
164.702 |
-9 |
-6.9 |
| Lumacaftor |
DB09280 |
152.924 |
-9 |
-7.1 |
| Naldemedine |
DB11691 |
159.413 |
-8.9 |
-7.8 |
| Etoposide |
DB00773 |
138.726 |
-8.9 |
-6.9 |
| Venetoclax |
DB11581 |
169.481 |
-8.8 |
-7.9 |
The values of binding energy for the interaction of deslanoside with Mproand ACE2 receptors were -9.3 and -9 kcal/mol, respectively. The binding models for the interaction of deslanoside with Mproand ACE2 are presented in Figures 1 and 2. It is noteworthy that the interactions of deslanoside with Mproand ACE2 receptors included hydrophobic interaction and hydrogen bonding. It is interesting that in the interaction of deslanoside with ACE2, hydrophobic forces were more important than hydrogen bonds.

Figure 1.
Docking Interaction of Deslanoside with a) Mpro and b) Interacting Amino Acids of Mpro. The interactions of Deslanoside with Mpro include hydrophobic interaction and hydrogen bonding.
.
Docking Interaction of Deslanoside with a) Mpro and b) Interacting Amino Acids of Mpro. The interactions of Deslanoside with Mpro include hydrophobic interaction and hydrogen bonding.

Figure 2.
Docking Interaction of Deslanoside with (a) ACE2 and (b) Interacting Amino Acids of ACE2. The interactions of Deslanoside with ACE2 receptor include hydrophobic interaction and hydrogen bonding. However, hydrophobic forces are more important than hydrogen bonds.
.
Docking Interaction of Deslanoside with (a) ACE2 and (b) Interacting Amino Acids of ACE2. The interactions of Deslanoside with ACE2 receptor include hydrophobic interaction and hydrogen bonding. However, hydrophobic forces are more important than hydrogen bonds.
Evaluation of Derivatives of 5-Hydroxy Indanone
According to the fragment-based QSAR model calculations, the percentage of inhibition of the activity of 5-hydroxy indanone derivatives against the ACE2 was predicted (Table 2). According to Table 2, compounds with an inhibition percentage above 45% entered the computational phase of docking and their energies of interaction with Mpro and ACE2 were calculated.
Table 2.
The Structure of 5-Hydroxy Indanone Derivatives, Experimental and Predicted Inhibition, and the Binding Energy for ACE2 and Mpro
|
|
|
ID
|
R
|
Inhibition (Exp)
|
Inhibition (Pred)
|
The Binding Energy for ACE2 (kcal/mol)
|
The Binding Energy for M
pro
(kcal/mol)
|
| 1 |
|
38.1188 |
37.1657 |
- |
- |
| 2 |
|
45.0495 |
44.9556 |
-6.8 |
-7.1 |
| 3 |
|
19.8019 |
19.2390 |
- |
- |
| 4 |
|
20.7920 |
18.5434 |
- |
- |
| 5 |
|
38.6138 |
37.1657 |
- |
- |
| 6 |
|
21.7821 |
22.9533 |
- |
- |
| 7 |
|
56.4356 |
56.4356 |
-7.5 |
-8.9 |
| 8 |
|
32.6732 |
25.6496 |
- |
- |
| 9 |
|
28.7128 |
37.6587 |
- |
- |
| 10 |
|
47.5247 |
49.1748 |
-6.8 |
-8 |
|
|
| 11 |
|
29.2682 |
28.9868 |
- |
- |
| 12 |
|
41.4634 |
39.3586 |
- |
- |
| 13 |
|
1 |
1 |
- |
- |
| 14 |
|
65.8536 |
67.9584 |
- |
- |
| 15 |
|
34.1463 |
34.3339 |
- |
- |
| 16 |
|
1 |
1.9830 |
- |
- |
| 17 |
|
43.9024 |
47.1845 |
- |
- |
| 18 |
|
78.0487 |
71.2027 |
- |
- |
| 19 |
|
46.3414 |
47.1845 |
- |
- |
| 20 |
|
56.0975 |
59.1936 |
- |
- |
| 21 |
|
100 |
99.2984 |
-6.6 |
-7.8 |
| 22 |
|
100 |
100 |
-6.6 |
-7.8 |
Indanone derivatives were selected as model descriptors. According to the model calculations, the linear method was selected for the relationship between descriptors and inhibition percentage. Thirteen fragments were selected as important and influential variables (Table 3). The statistical parameters of the model included R2=0.98, Q2LOO=0.94, and RMSD=7.12. Finally, considering the selected compounds in Table 2 and the fragments in Table 3, a new derivative with optimal properties was proposed (Figure 3).
Table 3.
The List of Descriptors (Fragments)
|
No
|
Fragments
|
| 1 |
C-O-C-C-H |
| 2 |
C-C-C=C-H |
| 3 |
H-C-C=C-C-O |
| 4 |
C-C=C-N-C-H |
| 5 |
H-C=C-C=C-N |
| 6 |
H-C-C=C-C-N=N |
| 7 |
C-C-C-C-C=C-H |
| 8 |
H-C-C-C-C-C-H |
| 9 |
C-C-C=C-C-C=C |
| 10 |
C=N-C-C=C-H |
| 11 |
C-C-C-N-C-C-N |
| 12 |
C-N-C-C=C-C-H |
| 13 |
F-C-C=C-C-H |
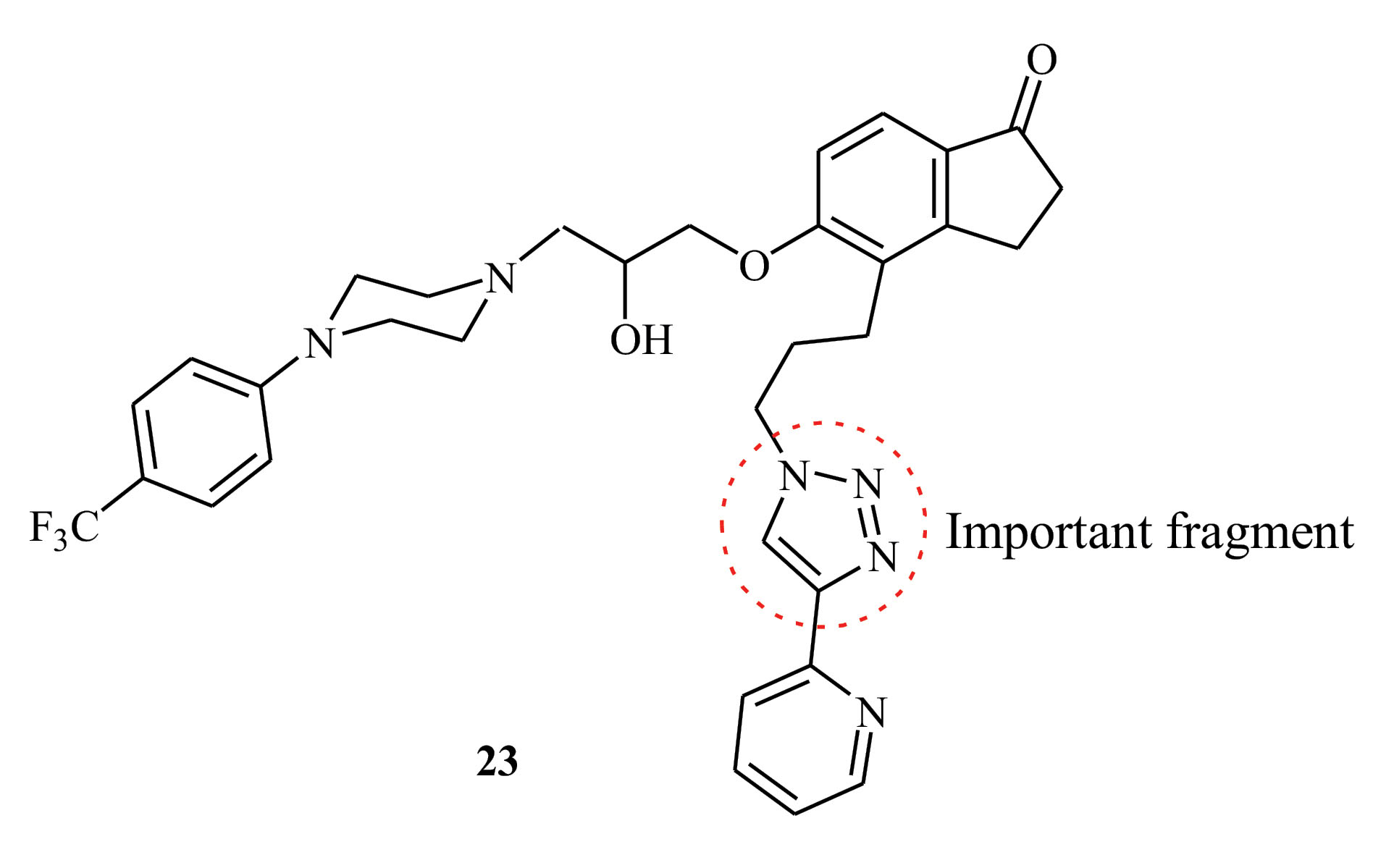
Figure 3.
Structure of Derivative 23.This derivative can be evaluated as a proposed compound in subsequent studies of its synthesis and activity against COVID-19.
.
Structure of Derivative 23.This derivative can be evaluated as a proposed compound in subsequent studies of its synthesis and activity against COVID-19.
Discussion
MproandACE2 haveessential role in SARS-CoV-2 replication process and entrance of the virus into the host cell (14,15). In this study, due to the critical role of Mpro and ACE2 in virus entrance into the host cell and virus replication, molecular docking and fragment-based QSAR method were used to discover the putative inhibitors of Mpro and ACE2.
The virtual screening of the approved drugs showed that deslanoside (DB01078) can interact effectively with both Mproand ACE2. Due to the molecular structure of deslanoside, the presence of hydroxy groups causes electrostatic interactions with Mproand ACE2 amino acids. However, the presence of interlocking six-membered and five-membered rings creates a hydrophobic nature and enables lipophilic interactions. The structural similarity of deslanoside to cholesterol well justifies hydrophobic interactions.
It is noteworthy that in recent years, researchers have designed and synthesized very suitable candidate compounds to inhibit the activity of ACE targets. These compounds, with a high capacity to inhibit ACE, can be good candidates for inhibiting COVID-19 activity.
In 2018, Vulupala et al identified 5-hydroxy indanone derivatives as ACE inhibitors. Their selected compounds included derivatives 2, 7, 10, 21, and 22 as the strongest ACE inhibitors (13). According to Table2, the predicted inhibition values are very similar to the experimental results found by Vulupala et al. Therefore, the experimental and predicted results were highly correlated (R2=0.98). According to the results of QSAR calculations, the 13 main fragments played a decisive role in the inhibitory activity of the studied compounds. Depending on the type of fragments and the presence of nitrogen atoms in most of them, the key role of triazole can be found. Triazole rings provide strong hydrogen bonds and strong electrostatic interactions with a variety of targets and are important in the drug design process (14).
According to the results of Table 2 and its comparison with the results of Table 1, it can be seen that compound 7 has optimal amounts of energy for interacting with ACE2 and Mpro. However, the energy value of deslanoside is more favorable than that of compound 7. It is noteworthy that derivatives 21 and 22 have a 100% inhibitory activity; therefore, their structural fragments are significant. Finally, considering the structure of derivatives 7, 21, and 22 and the fragments in Table 3, a new derivative with optimal properties can be proposed. Considering the main structure of derivatives 1-22 and all the mentioned parameters, derivative 23 can be evaluated as a proposed compound in subsequent studies of its synthesis and activity against COVID-19.
Conclusion
Our results showed that deslanoside and compound 23 potentially inhibit the activity of Mpro and suppress the essential replication process of the COVID-19 by interfering with the polyprotein processing. On the other hand, deslanoside can interact with the S protein binding site on ACE2 receptor, thereby preventing the recognition of the virus and its entrance into the host cell. The results of this study showed that the selected compound 23 is a good candidate for the treatment of COVID-19. However, further experimental validation is needed before conducting clinical trials.
Authors’ Contributions
SA, AB, and MS. contributed to the design and implementation of the research, to the analysis of the results and to the writing of the manuscript.
Conflict of Interest Disclosures
The authors declare that they have no conflict of interests.
Ethical Issues
The study was approved by the Ethics Committee of Hamadan University of Medical Sciences (No.: IR.UMSHA.REC.1399.120).
Acknowledgments
The study was funded by Vice-chancellor for Research and Technology, Hamadan University of Medical Sciences, Hamadan, Iran.
Funding
The study was funded by Vice-chancellor for Research and Technology, Hamadan University of Medical Sciences, Hamadan, Iran (No. 990209653).
References
- Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020; 368(6489):409-12. doi: 10.1126/science.abb3405 [Crossref] [ Google Scholar]
- Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov 2020; 6:14. doi: 10.1038/s41421-020-0153-3 [Crossref] [ Google Scholar]
- Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID-19: their roles in pathogenesis. J Microbiol Immunol Infect 2020. doi: 10.1016/j.jmii.2020.03.022 [Crossref]
- Kandeel M, Ibrahim A, Fayez M, Al-Nazawi M. From SARS and MERS CoVs to SARS-CoV-2: moving toward more biased codon usage in viral structural and nonstructural genes. J Med Virol 2020; 92(6):660-6. doi: 10.1002/jmv.25754 [Crossref] [ Google Scholar]
- Satarker S, Nampoothiri M. Structural proteins in severe acute respiratory syndrome coronavirus-2. Arch Med Res 2020; 51(6):482-91. doi: 10.1016/j.arcmed.2020.05.012 [Crossref] [ Google Scholar]
- Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 2020; 46(4):586-90. doi: 10.1007/s00134-020-05985-9 [Crossref] [ Google Scholar]
- Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020; 582(7811):289-93. doi: 10.1038/s41586-020-2223-y [Crossref] [ Google Scholar]
- Luan J, Lu Y, Jin X, Zhang L. Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem Biophys Res Commun 2020; 526(1):165-9. doi: 10.1016/j.bbrc.2020.03.047 [Crossref] [ Google Scholar]
- Robson B. Computers and viral diseases Preliminary bioinformatics studies on the design of a synthetic vaccine and a preventative peptidomimetic antagonist against the SARS-CoV-2 (2019-nCoV, COVID-19) coronavirus. Comput Biol Med 2020; 119:103670. doi: 10.1016/j.compbiomed.2020.103670 [Crossref] [ Google Scholar]
- Marinho EM, Batista de Andrade Neto J, Silva J, Rocha da Silva C, Cavalcanti BC, Marinho ES. Virtual screening based on molecular docking of possible inhibitors of Covid-19 main protease. Microb Pathog 2020; 148:104365. doi: 10.1016/j.micpath.2020.104365 [Crossref] [ Google Scholar]
- Vardhan S, Sahoo SK. In silico ADMET and molecular docking study on searching potential inhibitors from limonoids and triterpenoids for COVID-19. Comput Biol Med 2020; 124:103936. doi: 10.1016/j.compbiomed.2020.103936 [Crossref] [ Google Scholar]
- Abo-Zeid Y, Ismail NSM, McLean GR, Hamdy NM. A molecular docking study repurposes FDA approved iron oxide nanoparticles to treat and control COVID-19 infection. Eur J Pharm Sci 2020; 153:105465. doi: 10.1016/j.ejps.2020.105465 [Crossref] [ Google Scholar]
- Vulupala HR, Sajja Y, Bagul PK, Bandla R, Nagarapu L, Benerjee SK. Potent ACE inhibitors from 5-hydroxy indanone derivatives. Bioorg Chem 2018; 77:660-5. doi: 10.1016/j.bioorg.2018.02.022 [Crossref] [ Google Scholar]
- Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 2020; 46(4):586-90. doi: 10.1007/s00134-020-05985-9 [Crossref] [ Google Scholar]
- Keretsu S, Bhujbal SP, Cho SJ. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci Rep 2020; 10(1):17716. doi: 10.1038/s41598-020-74468-0 [Crossref] [ Google Scholar]