Avicenna Journal of Medical Biochemistry. 12(2):77-92.
doi: 10.34172/ajmb.2546
Original Article
Uncovering Potential Novel Antidiabetic Compounds from African Traditional Medicinal Plants: A Computer-Aided Study
Chimaobi James Ononamadu 1, *  , Mohnad Abdalla 2, Godwin Okwudiri Ihegboro 1
, Mohnad Abdalla 2, Godwin Okwudiri Ihegboro 1
Author information:
1Department of Biochemistry and Forensic Science, Faculty of Science, Nigeria Police Academy, Wudil-Kano, Nigeria
2Department of Pharmaceutics (Key Laboratory of Chemical Biology), School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Shandong Province, 250012, PR China
Abstract
Background: The prevalence of type 2 diabetes mellitus (T2DM) has increased markedly in recent years. Although traditional medicinal plants and natural products offer promising candidates for antidiabetic drugs, their full potential remains largely underexplored.
Objectives: This study aimed to identify antidiabetic phytocompounds from a database of African plant-derived compounds, which were screened against four key antidiabetic targets: alpha-amylase 1 (AMY1A), α-glucosidase (MGAM), Protein Tyrosine Phosphatase 1B (PTP1B), and dipeptidyl peptidase IV (DPP-IV).
Methods: The compounds were initially filtered for drug-likeness and subsequently screened using molecular docking. The top candidates underwent molecular dynamics (MD) simulations. During these simulations, the binding energies were calculated using the Molecular Mechanics Generalized Born Surface Area (MMGBSA) method. Additionally, several structural parameters such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (rGyr), polar surface area (PSA), molecular surface area (MolSA), and solvent accessible surface area (SASA) were analyzed.
Results: A total of 43 unique compounds belonging to several chemical classes (i.e., flavonoids, terpenoids, alkaloids, iridoids, and xanthones) were identified, exhibiting docking scores comparable to known controls. The results were as follows: docking scores of -7.4 to -8.7 kcal/mol (control: -9.7) for AMY1A, -6.8 to -8.0 kcal/mol (control: -8.2) for MGAM, -8.1 to -9.6 kcal/mol (control: -9.3) for DPP4, and -5.9 to -6.8 kcal/mol (control: -9.1) for PTP1B. MD simulations indicated that AMY1A-101679366 and DPP4-393472 complexes are negative and notably lower (-65.3 kcal/mol and -54.1 kcal/mol, respectively) than their respective controls. Furthermore, the MD simulations revealed relatively stable RMSD and RMSF profiles for the complexes, with fluctuations below 2.0 Å. The rGyr, PSA, MolSA, and SASA analyses further confirmed the stability of the protein-ligand complexes.
Conclusion: The findings unveiled several compounds with promising antidiabetic potential, establishing a basis for further in vitro and in vivo studies to explore their therapeutic applications in T2DM treatment. Additionally, these compounds may serve as scaffolds for enhanced drug development.
Keywords: Africa, Medicinal plants, Diabetes mellitus, Molecular dynamics, Molecular docking, Hypoglycaemic agents,
Copyright and License Information
© 2024 The Author(s); Published by Hamadan University of Medical Sciences.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium provided the original work is properly cited.
Please cite this article as follows: Ononamadu CJ, Abdalla M, Ihegboro GO. Uncovering potential novel antidiabetic compounds from African traditional medicinal plants: a computer-aided study. Avicenna J Med Biochem. 2024; 12(2):77-92. doi:10.34172/ajmb.2546
Background
Diabetes mellitus (DM) is a non-communicable disorder that has emerged as a leading global health concern (1,2). It is a metabolic disorder affecting carbohydrate, protein, and lipid metabolism, and is primarily characterized by chronic hyperglycaemia (3). The two major types of diabetes, type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM), are generally encountered in clinical practice, along with a small percentage of other forms of the disease. Type 2 diabetes accounts for more than 90% of the cases (4) and is characterized by a combination of insulin resistance and an inability to adequately compensate for insulin secretion actions and response (5). In contrast, type 1 results from an absolute deficiency in insulin secretion, necessitating insulin for management (4).
Studies from four decades ago reported a relatively low prevalence of DM, ranging from 0.8% to 2.5%. However, recent research indicates a significant rise in the global prevalence of DM, especially across Asia and Africa (6). According to the International Diabetes Federation (IDF) 2021 estimates, approximately 537 million people are currently living with diabetes, a 15.98% increase from the 463 million estimated in 2019 (7,8). Projections suggest that this number could rise to 783 million by 2045 if substantial efforts are not made to counteract the trend. Currently, about 537 million adults aged 20–79 are living with DM, with 75% of cases occurring in low- and middle-income countries. It is further predicted that the number of affected individuals could reach 643 million by 2030 and 783 million by 2045.
Poorly managed diabetes and chronic hyperglycaemia can lead to severe complications such as chronic renal failure, retinopathy, neuropathy, myocardial infarctions, gangrene, vascular damage, and other cardiovascular and metabolic comorbidities (9). Therefore, maintaining optimal blood glucose levels is crucial. Currently, there is a range of pharmacological options for treating and managing DM, including sulfonylureas, meglitinides, biguanides, thiazolidinedione, carboxylic enzymes inhibitors, incretins, sodium-glucose co-transporter 2 (SGLT2) inhibitors (10). Although most of these commercially available synthetic drugs have shown varying and significant degrees of efficacy in improving diabetic outcomes, literature reports indicate significant side effects and suboptimal activities across different brands. This underscores the need to intensify research efforts aimed at developing more efficient antidiabetic drugs with improved efficacy and minimal or tolerable side effects.
Historically, plants have been a vital source of nutrition and medicine (11). Plant-derived products offer numerous natural treatment options for diabetes, with over 800 medicinal plants known for their antidiabetic properties (12,13). These natural remedies are typically less expensive and have fewer side effects compared to synthetic drugs. Natural products are rich in stereogenic centers and occupy unique chemical spaces, making them valuable for developing safer and more effective antidiabetic agents (14). While research in this area has increased, many promising plant-based studies remain at the preliminary screening stage due to high costs and methodological challenges.
Advancements in analytical techniques like liquid chromatography-nuclear magnetic resonance (LC-NMR), liquid chromatography-mass spectrometry (LC-MS), gas chromatography-mass spectrometry (GC-MS), thin layer chromatography-mass spectrometry (TLC-MS), and computer-aided drug design have revolutionized drug discovery. In silico models, which screen and optimize potential drug-like molecules, are increasingly popular (15). These models can rapidly screen large numbers of molecules, significantly reducing the time and cost compared to traditional wet chemistry approaches. For instance, an in-silico model can screen 30 000 molecules in just five minutes, whereas the same task would take over 20 weeks using wet chemistry. Currently, (with in silico techniques) it is possible to almost simultaneously resolve a complex matrix of crude extract partially, identify potentially active compounds, and prioritize the identified compounds via virtual screening.
Identifying the exact pharmacological medication targets for the treatment of a disease is a critical component of drug development and discovery. The integration of the in silico technique into this process is largely dependent on the availability of a well-resolved protein target sequence or structural data. The completion of the human genome project, coupled with advancements in high-throughput protein purification, crystallography, and nuclear magnetic resonance spectroscopy techniques has resulted in several new therapeutic targets for drug discovery, with detailed structural data now readily accessible (16).
For antidiabetic treatments, potential targets could include any one or combinations of the following proteins: insulin/β-cell receptors, dopamine D2 receptor, nuclear receptors (e.g., PPAR-γ), enzymes involved in gluconeogenesis, carboxylic enzymes, dipeptidyl peptidase IV (DPP4), and glucagon-like peptide 1 (GLP-1), SGLT2, protein tyrosine phosphatase 1-Beta (PTP-1B), and glucokinase (17)
Virtual screening is a more direct and rational drug discovery approach that can be classified into ligand-based or structure-based methods. When a set of active ligand molecules is known, but no structural data is available for the target, ligand-based methods such as pharmacophore modeling and quantitative structure-activity relationship are used. Conversely, when adequate structural information is available for the target, structure-based methods are utilized, with molecular being the most common technique (16). Molecular docking is a computational algorithm used to model the interaction between a small molecule (ligand) and a known protein target. It involves predicting the ligand’s conformation, position, and orientation within the binding pocket or active site of the target protein. The nature and strength of the interactions are characterized quantitatively using some scoring functions. This approach can be used to explore and prioritize phytochemicals from plant extracts for further exhaustive studies. Several antidiabetic studies have demonstrated significant antioxidant and antidiabetic activity in plant extracts. While many of these plants have been profiled for their phytochemistry, only a few have been thoroughly explored to discover novel antidiabetic compounds. This study aimed to address this gap by identifying and screening African plant-derived phytocompounds for antidiabetic potential against four key antidiabetic targets using in silico techniques, especially molecular docking and molecular dynamics (MD) simulations.
Materials and Methods
Materials
Protein Targets
The following antidiabetic targets were used for the study:
Human DPP4 (PDB ID: 4PNZ),
Human protein tyrosine phosphatase 1B (PTP1B) (PDB ID: 4Y14),
Human Maltase-Glucoamylase (MGAM) (PDB ID: 2QMJ),
Pancreatic amylase (AMY1A) (PDB ID: 2QV4).
Ligands
The ligands used in this study were derived from two sources (with duplicates removed):
Phytochemicals from the literature on African plants used in folkloric ethnomedicine with reports of antidiabetic activities.
Natural compounds from the African Natural Product Database (ANPDB) (http://african-compounds.org/anpdb).
Methods
Compound Selection From the African Natural Product Database
The ANPDB database contains 954 compounds. To streamline the screening process, we applied a molecular weight range of 180–600, hydrogen bond acceptors ≤ 10, and hydrogen bond donors ≤ 5, based on the drug-likeness rules of Lipinski, Ghose (Amgen), Veber (GSK), Egan (Pharmacia), and Muegge (Bayer) (18-22). These filters were used since the in vivo activities of the compounds were unknown. After filtering, 620 compounds were selected for further analysis.
Literature Search for Antidiabetic Plants and Phytochemical Profiles
A literature search was conducted using PubMed and Google Scholar to identify antidiabetic compounds from African plants. The search terms included “Nigerian Plants AND antidiabetic compounds.” The inclusion criteria required reports of antidiabetic activity associated with African (Nigerian) plants.
Relevant phytochemicals were extracted from the literature, yielding 120 compounds. The 3D structural data for these compounds were downloaded in SDF format from PubChem and imported into molecular operating environment (MOE), where they were converted into MOE-compatible molecules. After filtering for molecular weight and eliminating duplicates, 101 compounds remained.
Given that many natural products with significant biological activity do not always conform to Lipinski’s rule of five (23-25), compounds that did not meet at least one parameter were still included to allow for more flexibility. This decision was based on reports suggesting that some active natural products exceed the typical limits for molecular weight, hydrogen bond donors, or acceptors. In total, 720 compounds were selected for computational screening.
All 3D ligand files were organized into an mdb database in MOE for docking.
Molecular Docking Simulation
Protein Target Preparation
The x-ray crystallography structural data for each of the selected important targets- PTP1B (4Y14), DPP4 (4PNZ), AMY1A (2QV4), and MGAM (2QMJ)- were downloaded in PDB format from the Research Collaboratory for Structural Bioinformatics (RCSB) PDB database (http://www.rcsb.org/pdb), along with their respective co-crystallized ligands, which served as controls. The preparation and minimization of the structures were performed using tools and protocols in MOE, as previously described (26). The preparatory process included the removal of water molecules and other co-crystalized molecules, protonation and application of partial charges, and energy minimization, which was implemented using the QuickPrep function in MOE. Then, the fully prepared and optimized 3D structure was saved in MOE format for use in docking simulations.
Binding/Docking Site Prediction
The binding site for molecular docking was defined by the co-crystalized ligand bound to the target protein. The ligand binding site option was selected as the docking site during the docking simulation in MOE.
Validation of Molecular Operating Environment Docking Program
A preliminary docking study was conducted using the X-ray crystallography structure data of the target proteins and their co-crystallized ligands. The co-crystallized ligand was extracted and re-docked into the binding site of each target protein. This process was repeated using various scoring functions each time, including ASE, Affinity dG, Alpha HB, Electron Density, GBVI/WSA dG, and London dG. The docked binding pose for each scoring function was then compared to the experimentally determined pose in the crystal structure. A root mean square deviation (RMSD) value of ≤ 2.0 Å, relative to the native binding pose of the control ligand, was considered a successful outcome, validating the docking software (27). The combination of London dG and GBVI/WSA dG scoring functions produced docking poses within 2.0 Å of the experimentally determined pose (see Table S1) for all four target proteins used in the study. Therefore, the default scoring function of London dG/GBVI/WSA dG was selected for further use in this study.
Docking Simulation
Docking simulations were performed using MOE. The ligand was selected and docked using the Triangular Matcher/rigid receptor method and scored using London dG/GBVI/WSA options on a system with an Intel Core i7 CPU @ 2.00 GHz, 2.60 GHz. The Triangular Matcher method (default in MOE) is regarded as the most effective placement method for standard and well-defined binding sites in MOE (28). It generates poses by superimposing triplets of ligand atoms with triplets of receptor sites (alpha centers, which represent locations of tight packing) (28). The poses generated by the placement method were scored using the London dG scoring function and subsequently re-scored using GBVI/WSA dG.
The protein-ligand docking poses and their respective scores were saved in a database in mdb format, and the interactions between the ligands and target proteins were visualized (both 2D and 3D) using Discovery Studio and MOE’s ligand interaction options. The docking scores for the top-ranking poses of each key compound were normalized by computing their corresponding size-independent ligand efficiencies (SILEs) (29) using the following formula (Supplementary file 1, Tables S2-S5)
where D is the docking binding energy score, and N is the number of heavy atoms in the ligand.
Identification of Potentially Multitargeting Compounds
To identify promising compounds with potentially multimodal mechanisms of action, multitargeting compounds were selected from the list of high-scoring docked compounds using Venny2.1 (https://bioinfogp.cnb.csic.es/tools/venny/).
Molecular Dynamics Simulation
Four of the highest-ranking docked protein-compound complexes (at least one from each category) were selected for a 100 ns MD simulation using Schrodinger’s Desmond module using the OPLS2005 forcefield, as described by (30). This simulation was performed on a high-performance computing system equipped with dual Intel Xeon Gold 6248R processors (48 cores total), two NVIDIA Quadro RTX 6000 GPUs (24 GB VRAM each), 128 GB of DDR4 ECC RAM, and a storage setup consisting of 2 TB NVMe and 4 TB SATA SSDs. The solvated water-soaked system was created using the Desmond System Builder tool and the TIP3P solvation model. An orthorhombic box was simulated with a boundary distance of at least 10 Å from the protein’s outer surface, using periodic boundary conditions. The system was neutralized with the addition of 0.15 M NaCl to maintain isosmotic conditions. A pre-defined equilibration procedure was performed before the simulation. The MD simulation was conducted at a pressure of 1.0 bar and a temperature of 300 K (considering the target protein and organism are insects, not humans), with 1000 frames saved to the trajectory over the 100 ns period. The trajectory file of the simulated system was then used to calculate various structural parameters such as RMSD, root mean square fluctuations (RMSF), radius of gyration (rGyr), protein-ligand contacts, intermolecular hydrogen bonding (H-bonding), solvent-accessible surface area (SASA), molecular surface area (MolSA), and polar surface area (PSA) (30).
Binding Energy Calculation
The docked conformations were energy-minimized using the Prime module of Schrodinger, and then the molecular mechanics generalized born surface area (MM-GBSA) analysis was implemented to calculate the binding free energy of the complexes (31).
The formula is given below:
ΔG(bind) = ΔG(solv) + ΔE(MM) + ΔG(SA)
where ΔGsolv is the difference in GBSA solvation energy between the protein-ligand complex and the sum of the solvation energies for unliganded protein and ligand. ΔEMM is the difference in the minimized energies between the protein-ligand complex and the sum of the energies of the unliganded protein and ligand. ΔGSA is the difference in surface area energies of the complex and the sum of the surface area energies for the unliganded protein and ligand.
Results
DM remains a significant global health issue, with rising prevalence and associated complications that underscore the urgent need for new therapeutic options. While current treatments are available, the search for novel therapies continues, with natural products emerging as promising sources of antidiabetic compounds. Africa, renowned for its rich biodiversity and long-standing cultural tradition of plant use, is home to a vast heritage of medicinal plants traditionally used to treat diabetes. Although there is no comprehensive repository of these plants, the literature is replete with studies validating their antidiabetic properties through experimental investigations.
For instance, Mohammed et al (32) conducted an extensive review of in vivo antidiabetic studies from 2000 to 2013, reporting that the Asteraceae and Lamiaceae families are the most frequently reported. Similarly, Mohammed et al (33) reviewed 18 African medicinal plants and one herbal product tested for antidiabetic activity in human subjects, finding that extracts from Laportea ovalifolia and Momordica charantia are particularly effective in managing fasting blood glucose, without showing toxicity, regardless of study duration or dosage. This was further supported by van de Venter et al (34) who evaluated and scored 11 South African plants and identified Brachylaena discolor as highly effective.
Pereira et al (35) used computational approaches to identify 430 compounds from 184 African plants as potential inhibitors of antidiabetic targets. New sources of bioactive compounds with antidiabetic potential included Argemone ochroleuca, Clivia miniata, Crinum bulbispermum, Danais fragrans, Dioscorea dregeana, Dodonaea angustifolia, Eucomis autumnalis, Gnidia kraussiana, Melianthus comosus, Mondia whitei, Pelargonium sidoides, Typha capensis, Vinca minor, Voacanga africana, and Xysmalobium undulatum.
Despite these promising findings, most of these plants have not been fully characterized regarding their active compounds and mechanisms of action, highlighting the need for further research to advance drug discovery.
This study aimed to identify potential antidiabetic agents from various plant sources using computational docking simulations. Enzymes such as AMY1A and MGAM have gained popularity in managing DM and obesity due to their role in reducing postprandial sugar levels by inhibiting carbohydrate-metabolizing enzymes (36,37). Additionally, PTP1B is a critical target for enhancing insulin sensitivity as it acts as a negative regulator of insulin and leptin signaling pathways, playing a major role in insulin desensitization (38). On the other hand, DPP4 is targeted to extend the half-life of incretins, thereby maintaining glucose homeostasis by increasing insulin secretion and reducing glucagon secretion (39,40).
In the study, a virtual library of 720 compounds was screened against four key antidiabetic targets (i.e., Human AMY1A, Human MGAM, Human PTP1B, and Human DPP4 using molecular docking, which yielded 39 unique compounds. The results of the molecular docking simulation are presented in Tables 1-4, Figure 1 and Figure S1. The top 12 compounds, ranked by their SILE scores, are presented in Tables 1-4 (Figure S2) along with their docking scores and interactions with the target amino acid residues. The compounds showing potential for multiple targets are presented in a Venn diagram (Figure 1). The results revealed that the docking simulations identified several compounds with docking energy scores and SILE values comparable to those of the control. These compounds belong to various chemical classes, including flavonoids, terpenoids, alkaloids, iridoid/secoiridoid glycosides, and xanthones.
The stability of some of these compounds in complex with their respective targets was further investigated using molecular dynamics simulations. The results are presented in Figures 2-6 and Figures S2-S7, showing the MMGBSA-computed binding energies, root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (rGyr), hydrogen bonding interactions, solvent-accessible surface area (SASA), molecular surface area (MolSA), and polar surface area (PSA).
Discussion
Molecular docking
Pancreatic α-Amylase
For AMY1A (Table 1), the top-scoring compound types included hypoxoside, licarin-type phenylpropanoid, barbigerone-type pyrano-isoflavone, acetogenin, cucurbitacin-type steroidal diterpene, isoquinoline alkaloid, microfolian, calceolarioside-type hydroxy cinnamic acid glycosides, rutin, kulonate-type diterpene, vicenin, and quercetin. The docking binding energies of these compounds ranged from -7.443 to -8.657 kcal/mol, compared to the control at -9.670 kcal/mol. SILE values ranged from 2.251 to 2.821, with the control value at 2.906.
Table 1.
Docking Binding Energies and Size-Independent Ligand Efficiencies of the Top-ranked Compounds Docked in Human Pancreatic Amylase (PDB ID:2QV4.)
|
S/N
|
Compound Description
(PubChem ID)
|
Docking Energy (SD)
|
SILE
|
Interacting Amino Acids
|
| 1 |
Hypoxoside (101679366) |
-8.657 (0.323) |
2.821 |
(Asp197, Asn53, Asp300, Glu233)a, Asp197, Asp300, Asn53)b, Trp59f, (Trp59, Asp300)e |
| 2 |
Licarin-type phenylpropanoid (132989152) |
-7.725 (0.643) |
2.813 |
(Asp300, Trp59, Glu240)b, (Glu233, Ile235)f, |
| 3 |
Barbigerone-type pyranoisoflavone (132989074) |
-7.719 (0.262) |
2.811 |
(Asp300, His305)b, (Glu233, Ile235,Tyr62)c (lys200, Ile235, Trp59, His 201, His 305)e |
| 4 |
Acetogenin (393472) |
-7.886 (0.2613) |
2.763 |
(Tyr151, Asp300, His201)a, His305b, (Ile235, leu165, Trp59, Tyr62, His101)e |
| 5 |
Cucurbitacin-type steroidal diterpene (162944977) |
-8.077 (0.737) |
2.756 |
(Lys200, Asp197)a, His201b, (Leu162, Leu165,Trp59)e |
| 6 |
Emetine (Isoquinoline alkaloid) (10219) |
-7.917 (0.433) |
2.725 |
Lys200a, (Glu240, Asp300, Asp197, Trp58)b, Tyr151c, (Lys200, Ile235, Trp58, Trp59, His201, His305)e |
| 7 |
Microfolian (10884656) |
-7.637 (0.107) |
2.700 |
(His305, Asp300)a, Asp197b, (Trp59, Leu162, Ile235, Trp59, His201)e, |
| 8 |
Calceolarioside- type hydroxy cinnamic acid glycosides (132989156) |
-7.443 (0.433) |
2.630 |
(Lys200, Asp197, Glu233, Asp300)b, Gln63c, Ile235f |
| 9 |
Rutin (5280805) |
-8.101 (0.551) |
2.621 |
(Asp197, Glu233)a, (Asp300, Trp59), (Trp59, His305, Ile235)e |
| 10 |
Kulonate - type diterpene (163058659) |
-7.681 (0.0353) |
2.621 |
Asp300a, (Ile235, Leu163, Trp59, Tyr62, His305)e |
| 11 |
Vicenin (442664) |
-7.764 (0.1497) |
2.530 |
(Trp59, Glu233)a, (Asp197, Asp300)b, Tyr62c, (Ile235, Trp59)c |
| 12 |
Quercetin (5280343) |
-5.689 (0.634) |
2.251 |
(Glu233, Asp300)a, (Glu233,Asp300)d, Ile235f, leu162e |
| 13 |
Control (24755467) |
-9.670 (0.142) |
2.906 |
(Gly164, Gln63, Asp300, Trp69, Glu233, Gln63, His101, Arg195, His299, Tyr62, Asn105, Ala106, Val107, Leu162)a, (Asp300, Glu233, Asp197)d |
Note. SILEs: Size independent ligand efficiencies; SD: Standard deviation; Superscripts: a-Conventional H-bond, b-Carbon H-bond, c- Pi-donor H-bond, d-Electrostatic, e- Alkyl/pi-alkyl, f- other pi-bonds, g-Halogen.
The docked complexes were stabilized by a combination of polar and hydrophobic interactions. Most of the high-ranking compounds—101679366, 393472, 162944977, 10219, 10884656, 5280805, 163058659, 442664, and 5280343—were stabilized by 1-3 strong hydrogen bonds involving key active site amino acid residues such as Asp197, Asn53, Tyr151, Asp300, His201, Lys200, His305, Glu233, and Trp59.
These compounds are structurally diverse, encompassing flavonoids, complex polyphenolics, diterpenes, and alkaloids. Their structures, featuring multiple cyclic moieties (especially benzene rings) and hydroxylation patterns, facilitate the stabilization of the complexes. The hydroxyl groups frequently participate in hydrogen bonding with enzyme residues, while the aromatic rings allow for π-π stacking interactions.
Maltase-Glucoamylase
For MGAM (Table 2), several high-ranking compounds were identified, including swertiamarin, sweroside, baicalin, prenylated flavonoids (e.g., mundulinol and its hydroxylated derivative), loganin, lupinifolin, mundulin, microfolian, an acetylated flavonoid, and other simple flavonoids. The docking binding energies for these compounds ranged from -6.804 to -7.971 kcal/mol, compared to the control at -8.281 kcal/mol. SILE values ranged from 2.474 to 2.818, versus the control at 2.906. All the selected compounds fell into two structural categories: flavonoids (including xanthones) and iridoid monoterpenoids. With the exception of the acetylated flavonoid, all other compounds formed 2-5 strong hydrogen-bond interactions with key amino acid residues, including Arg526, Asp542, Asp327, His600, Thr205, Asp203, Asp443, and Thr544.
Table 2.
Docking Binding Energies and Size-Independent Ligand Efficiencies of the Top-Ranked Compounds Docked in Human Maltase-Glucoamylase (PDB ID:2QMJ.)
|
S/N
|
Compound Description
(PubChem ID)
|
Docking Score (SD)
|
SILE
|
Interacting Amino Acids
|
| 1 |
Baicalin (64982) |
-7.971 (0.143) |
2.818 |
(Arg526, Asp542, Asp327, His600)a, (Asp443, Asp542, Asp327)b, Trp406f |
| 2 |
Swertiamarin (a secoiridoid glycoside) (442435) |
-7.471 (0.377) |
2.811 |
(Thr205, Asp203, Asp542, Asp327)a, (Asp443, Asp327, Asp203, Asp327)b, Trp406e |
| 3 |
Sweroside (an iridoid glycoside) (161036) |
-7.176 (0.226) |
2.732 |
(Thr205, Arg526, Asp542, Asp443, Asp327)a, (Asp443, Asp327, Asp203)b, Tyr299f, Trp406e |
| 4 |
Prenylated flavonoid (132988919) |
-7.392 (0.146) |
2.566 |
(Asp443, Asp542)a, Asp443b, Asp542d, (Met444, Trp406)f, (Tyr299, Trp406, Phe575, His600, Tyr605)e |
| 5 |
Lupinifolinol (241097691) |
-7.170 (0.332) |
2.559 |
(Thr205, Arg526, Asp203, Thr544)a, Asp542b, Asp542d, Thr205c, Mett444f, (Met444, Ile328, Tyr299, Trp406, Phe575, His600, Ala576)e |
| 6 |
Mundulinol (10363971) |
-7.082 (0.238) |
2.553 |
(Thr205, Arg526, Asp203, Thr544)a, Asp542b, Asp542d, Thr205c, Mett444f, (Met444, Ile328, Tyr299, Trp406, Phe575, His600, Ala576)e |
| 7 |
Loganin (an iridoid monoterpenoid) (87691) |
-6.854 (0.058) |
2.550 |
(Asp443, Asp327)a,(Asp542, Asp203, Asp443, Asp327)b, Tyr299f, (Phe450, Phe575)e |
| 8 |
Lupinifolin (10250777) |
-7.051 (0.252) |
2.541 |
(Arg526, Asp203, Thr544)a, Asp542d, Thr205c, Met444f, (Met444, Ile328, Tyr299, Trp406, Phe575, His600, Ala576)e |
| 9 |
Flavonoid (163085068) |
-6.804 (0.121) |
2.531 |
(Arg526, Asp203, Thr205)a, Asp542d, (Ala576, Met444)f, (Ile328, Tyr299, Trp406, Trp441, Phe575)e |
| 10 |
Mundulin (15895372) |
-6.926 (0.116) |
2.522 |
(Arg526, Asp203)a, Asp542d, Thr205c, Met444f, (Met444, Ile328, Tyr299, Trp406, Phe575, His600, Ala576)e |
| 11 |
Acetylated flavonoid (163058856) |
-7.186 (0.642) |
2.495 |
Asp542d, (Tyr299, Trp406)f, (Ala576, Tyr299, Trp406)e |
| 12 |
Microfolian (10884656) |
-6.997 (0.096) |
2.474 |
(Arg526, Asp203)a, Asp542b, Lys480f, (Ile328, Met444, Tyr299, Trp406, Phe575, Tyr605, Lys480)e |
| 13 |
Control (445421) |
-8.281 (0.974) |
2.661 |
(Arg526, Asp203, Met444, Asp542, His600, Asp327, Thr205)a, (Asp542, Asp203, Asp443)d (Tyr299, Trp406, Phe575)e |
Note. SILEs: Size independent ligand efficiencies; SD: Standard deviation; Superscripts: a-Conventional H-bond, b-Carbon H-bond, c- Pi-donor H-bond, d-Electrostatic, e- Alkyl/pi-alkyl, f- other pi-bonds, g-Halogen.
The iridoid glycosides could bind effectively to the MGAM binding site, with complex stabilization achieved through hydrogen bonds involving multiple hydroxyl groups, as well as additional π interactions. The flavonoids and xanthones also interacted stably with critical amino acid residues within or near the substrate binding site. Their heterocyclic structures and diverse patterns of hydroxylation and prenylation contributed significantly to the stability and high binding energy scores.
Dipeptidyl Peptidase IV
For DPP4 (Table 3), the key compounds identified included acetogenin, severifoline-type acridone alkaloid, isodiscoloranone, obliquine alkaloid, discoloranone, a prenylated flavonoid, gartanin, corynantheine alkaloid, parviflorone-type diterpene, microfolian, flavonoid glycoside, and rutin. Docking binding energies for these compounds ranged from -8.143 to -9.759 kcal/mol, with the control at -9.725 kcal/mol. SILE values were between 2.566 and 3.419, compared to the control at 3.847.
Table 3.
Docking Binding Energies and Size-Independent Ligand Efficiencies of the Top-Ranked Compounds Docked in Human DPP4 (PDB ID:4PNZ)
|
S/N
|
Compound Description
(PubChem ID)
|
Docking Score (SD)
|
SILE
|
Interacting amino acids
|
| 1 |
Acetogenin (393472) |
-9.759 (0.262) |
3.419 |
(Ser209, Glu206, Glu205 Arg358)a, Ser209b, (Arg358, Val656, Phe357, Tyr662, Tyr666)e |
| 2 |
Severifoline- type Acridone alkaloid(163042290) |
-9.019 (0.551) |
3.159 |
(Tyr662, Glu206,)a, Val 207, Glu206, Ser209)b, Glu205d, (Arg358, Val711, Val656, Tyr547,Trp659, Tyr662, Tyr666)e |
| 3 |
Isodiscoloranone (132989036) |
-8.896 (0.633) |
3.116 |
(Arg358, Glu361)a, Val 207, Ser209, Arg358)b, Arg358c, Phe357f, Phe357, Tyr659, Tyr662,Tyr666,Arg358)e |
| 4 |
Obliquine alkaloid (162887088) |
-8.795 (1.181) |
3.081 |
Arg669a, Glu206b, Tyr547c, Phe357, Tyr666)f, (Phe357, Tyr547, Val656, Val711)e |
| 5 |
Discoloranone (11995377) |
-8.776 (0.395) |
3.074 |
(Ser209, Tyr666)a, (Glu206, Ser630)b, Phe357f, (Arg358, Val656, Val711, Phe357, Tyr662)e |
| 6 |
Prenylated flavonoid (162912953) |
-8.543 (0.239) |
3.049 |
(Tyr666a, Glu206, Ser630)b, (Arg358, Val656, Val711, Phe357, Tyr666)e |
| 7 |
Corynantheine alkaloid (3037997) |
-8.143 (0.400) |
3.029 |
(Glu206, Tyr666, Glu205, Tyr670, Tyr547)a, Tyr662c, (Phe357, Tyr662, Tyr666, Val711)e |
| 8 |
Gartanin (5281633) |
-8.288 (1.068) |
3.018 |
(Ser630, Tyr662, Glu206)a, Glu205d, (Val656, Val711, Tyr547,Trp659, Tyr662, Tyr666)e |
| 9 |
Flavonoid glycoside (163065490) |
-8.193 (0.253) |
3.015 |
(Glu206, Ser630, Tyr662, Asn710, Val207)a, (Glu206, Tyr662)b, Ser209c, Tyr631e |
| 10 |
Parviflorone-type diterpene (10366501) |
-8.217 (0.561) |
2.878 |
(Ser630, Glu206)a, Val207b, Tyr662c, Tyr666f, (Arg358, Phe357)e |
| 11 |
Rutin (5280805) |
-8.911 (0.233) |
2.883 |
(Glu206, Ser630, Tyr662, Asn710, Val207)a, (Glu206, Tyr662)b, Ser209c, Tyr631e |
| 12 |
Microfolian (10884656) |
-8.161 (0.416) |
2.866 |
Tyr547, Glu205)a, Glu206b, Tyr547f, (Val656, Val711, Val656, Phe357,Trp659, Tyr662, Tyr666)f |
| 13 |
Control (46209242) |
-9.725 (0.822) |
3.847 |
(Glu205, Glu206, Arg125, Tyr547, Asn710)a, Tyr662c, (Asp663, Tyr666)d, (Tyr662, Phe802, Tyr666)f, (Glu205, Asn710)g |
Note. DPP4: Dipeptidyl peptidase IV; SILEs: size independent ligand efficiencies; SD: Standard deviation; Superscripts: a-Conventional H-bond, b-Carbon H-bond, c- Pi-donor H-bond, d-Electrostatic, e- Alkyl/pi-alkyl, f- other pi-bonds, g-Halogen.
Structurally, these compounds are diverse, falling into five categories: polyketides, alkaloids, diterpenes, α-acids, and flavonoids. This structural diversity is reflected in their binding poses and interaction modes with DPP4 amino acid residues. Except for the acetylated flavonoid, all compounds demonstrated 2-5 stabilizing hydrogen bonds with residues such as Ser209, Glu206, Arg358, Tyr662, Glu361, Arg669, Tyr666, Glu205, Tyr670, Tyr547, Ser630, and Asn710. Each compound class contributes uniquely to binding and interaction with DPP4, as indicated by docking binding energy scores and SILE values, which were comparable to the control. Despite their structural diversity, all the compounds commonly feature multiple heterocyclic moieties, particularly benzene rings, along with varied hydroxylation patterns. These characteristics significantly enhance the binding affinity and stability of the complexes.
Protein Tyrosine Phosphatase 1B
For PTP1B (Table 4), the identified compounds included limonianin-type flavone, lupinifolinol, pilostigmol-type phenylpropanoid, rutin, acetogenin, diuvaretin (a hydrochalcone), barbigerone-type pyranoisoflavone, curcumin, parviflorone-type diterpene, luteolin-7-glucoside, and prenylated xanthones/flavonoids such as tovophyllin B and gartanin. These compounds fall into three main categories: flavonoids, phenylpropanoids, and diterpenes. The docking binding energies for these compounds ranged from -5.889 to -6.751 kcal/mol, whereas the control compound exhibited a binding energy of -9.098 kcal/mol. SILE values for these compounds ranged from 2.140 to 2.540, while the control’s SILE was 3.464.
Table 4.
Docking Binding Energies and Size-Independent Ligand Efficiencies of the Top-Ranked Compounds Docked in Human PTP1B (PDB ID:4Y14.)
|
S/N
|
Compound Description
(PubChem ID)
|
Docking Score (SD)
|
SILE
|
Interacting Amino Acids
|
| 1 |
Limonianin-type flavone (132988935) |
-6.751 (0.611) |
2.540 |
Asp48b, Tyr46c, Phe182f, (Ala217, Ile219, Cys215, Phe182, Ala217)e |
| 2 |
Lupinifolinol (241097691) |
-6.628 (0.194) |
2.366 |
Asp48a, Asp48d, Phe182f, Tyr46f, (Ala217, Ile219, Cys215, Phe182, Ala217) |
| 3 |
Pilostigmol-type phenylpropanoid (162856705) |
-6.245 (0.195) |
2.298 |
Asp48a, Asp48d, Phe182f, Tyr46f, (Met258, Tyr46, Ala217)e |
| 4 |
Tovophyllin B (Xanthone prenylated) (509268) |
-6.538 (0.1651) |
2.270 |
Asp181b, Asp48d, (Tyr46, Phe182)f, (Ala217, Ile219, Val49, Met258, Phe182)e |
| 5 |
Rutin (5280805) |
-6.987 (0.5046) |
2.261 |
(Asp181, Asp48, Gly262)a, (Ala217, Tyr46)f |
| 6 |
Acetogenin (393472) |
-6.434 (0.3055) |
2.254 |
(Ser216, Asp48)b, (Tyr46, Phe182)e |
| 7 |
Diuvaretin (Hydrochalcones) (3085222) |
-6.600 (0.2068) |
2.252 |
Asp48b, Asp48d, (Tyr46, Phe182)f, (Ala217, Val49, Tyr46, Ala217)e |
| 8 |
Barbigerone-type pyranoisoflavone (132989074) |
-6.162 (0.0961) |
2.244 |
Asp48b, Asp48d, Tyr46f, (Arg47, Ala217, Ile219,Cys215, Phe182)e |
| 9 |
Curcumin (969516) |
-6.007 (0.2056) |
2.235 |
(Asn44, Arg45, Gln262)b, Tyr46c, Tyr46f, (Ala217, Val 49, Ile219, Phe182, Arg47, Ala217)e |
| 10 |
Parviflorone-type diterpene (101967010) |
-6.182 (0.1055) |
2.186 |
Asp48d, Phe182f, (Tyr46, Ala217)e |
| 11 |
Luteolin-7-glucoside (5280637) |
-6.093 (0.2054) |
2.150 |
Arg221a, Asp48a, Tyr46c, (Ala217, Tyr46)f |
| 12 |
Gartanin (5281633) |
-5.889 (0.501) |
2.140 |
(Ala217, Tyr46, Phe182)f, Ala217e |
| 13 |
Control (91826021) |
-9.098 (0.215) |
3.464 |
(Arg221, Asp181)d, (Ser219 Ala217, Ile219, Gly220, Arg221, Asp48)a, Asp181g, (Ala217, Phe182)f. |
Note. PTP1B: Protein tyrosine phosphatase 1B; SILEs: Size independent ligand efficiencies; SD: Standard deviation; Superscripts: a-Conventional H-bond, b-Carbon H-bond, c- Pi-donor H-bond, d-Electrostatic, e- Alkyl/pi-alkyl, f- other pi-bonds, g-Halogen.
Relative to the control, these compounds showed notable differences in docking binding energies and SILE values. To explain the significant differences in binding energy, it is important to consider the nature of interactions formed between each compound and PTP1B’s amino acid residues. Most of these compounds engage in primarily hydrophobic interactions with PTP1B’s active site residues. A few compounds such as lupinifolinol, pilostigmol-type phenylpropanoid, rutin, acetogenin, and luteolin-7-glucoside formed 1-2 hydrogen bonds, mostly with residues Asp48 and Arg211. However, these interactions were relatively limited compared to the control. These limited interactions may have contributed to the observed higher binding energies (weaker binding) and lower SILE scores of these compounds compared to the control.
In contrast, the control compound, a synthetic experimental molecule with a central phenylalanine moiety and extended difluoro (phosphono) methyl and N-methyl-N-alpha (methylsulfonyl) side chains, is specifically designed to maximize binding stability. Its electronegative side chains facilitated extensive hydrogen bonding with PTP1B residues such as Arg219, Ala217, Ile219, Gly220, Arg221, and Asp48. This abundance of hydrogen bonds significantly enhanced binding stability, resulting in a lower binding energy (stronger binding) and a higher SILE score than the natural compounds that were screened.
Overall, the top-ranking compounds identified for the four targets belong to different structural categories. However, it can be inferred that structurally, the presence of a conjugated aromatic ring (heterocyclic rings in some cases), along with varied hydroxylation patterns, were key features contributing to their binding affinity and stability.
Previous in silico studies have identified various classes of compounds as effective inhibitors of diabetic targets. For instance, Nguyen et al (41) identified β-amyrin, taraxerol, 1-O-galloyl-β-D-glucose, corilagin, cosmosiin, quercetin-3-galactose, and quercitrin with high binding affinities for several diabetic targets, including PTP1B. In another in silico study, Damián-Medina et al (42) reported delphinidin-3-glucoside and petunidin-3-glucoside as potential PTP1B inhibitors among polyphenolics. Macalalad et al (43) also reported triterpenoids and diterpenoids as promising inhibitors of PTP1B, DPP4, SGLT-4, and Fructose-1,6-Bisphosphatase (FBPase), specifically identifying gypsogenin for PTP1B and adunctin for DPP-4.
Interestingly, some of the identified compounds or compound classes have been reported in vitro and in vivo studies to demonstrate significant antidiabetic activity or mechanisms, including barbigerone (44), curcubitacins (45,46), mangosteen xanthones (47), sweroside (48), swetiamarin (49), coumarins and acridone alkaloids (50), alkaloids (51,52), lupinifolin (53), rutin (54-56), quercetin (57), carpachromene (58).
Venn Diagram Identification of Potential Multitargeting Compound
A Venn diagram analysis revealed several compounds with potential multi-target activity such as gartanin, rutin, the barbigerone-type pyrano-isoflavone, microfolian, lupinifolinol, and acetogenin (Figure 1). The discovery of compounds with multi-target profiles is particularly promising as diabetes is a complex disease requiring multifaceted therapeutic interventions, warranting further investigation into their potential antidiabetic effects. This is also corroborated by some previous studies. Specifically, the multitarget inhibitory action of some prenylated xanthones has been reported against amylase, glucosidase, and PTP1B (59,60). Similarly, Quercetin has shown activity against DPP4 (61), PTP1B (62), amylase, and glucosidase (63), prenylated flavonoids against PTP1B (64), and lupinifolin against amylase and glucosidase (53). Additionally, rutin has shown inhibitory activity against PTP1B (65), amylase, and glucosidase (63,66), and barbigerone has been found to target amylase and glucosidase (67).
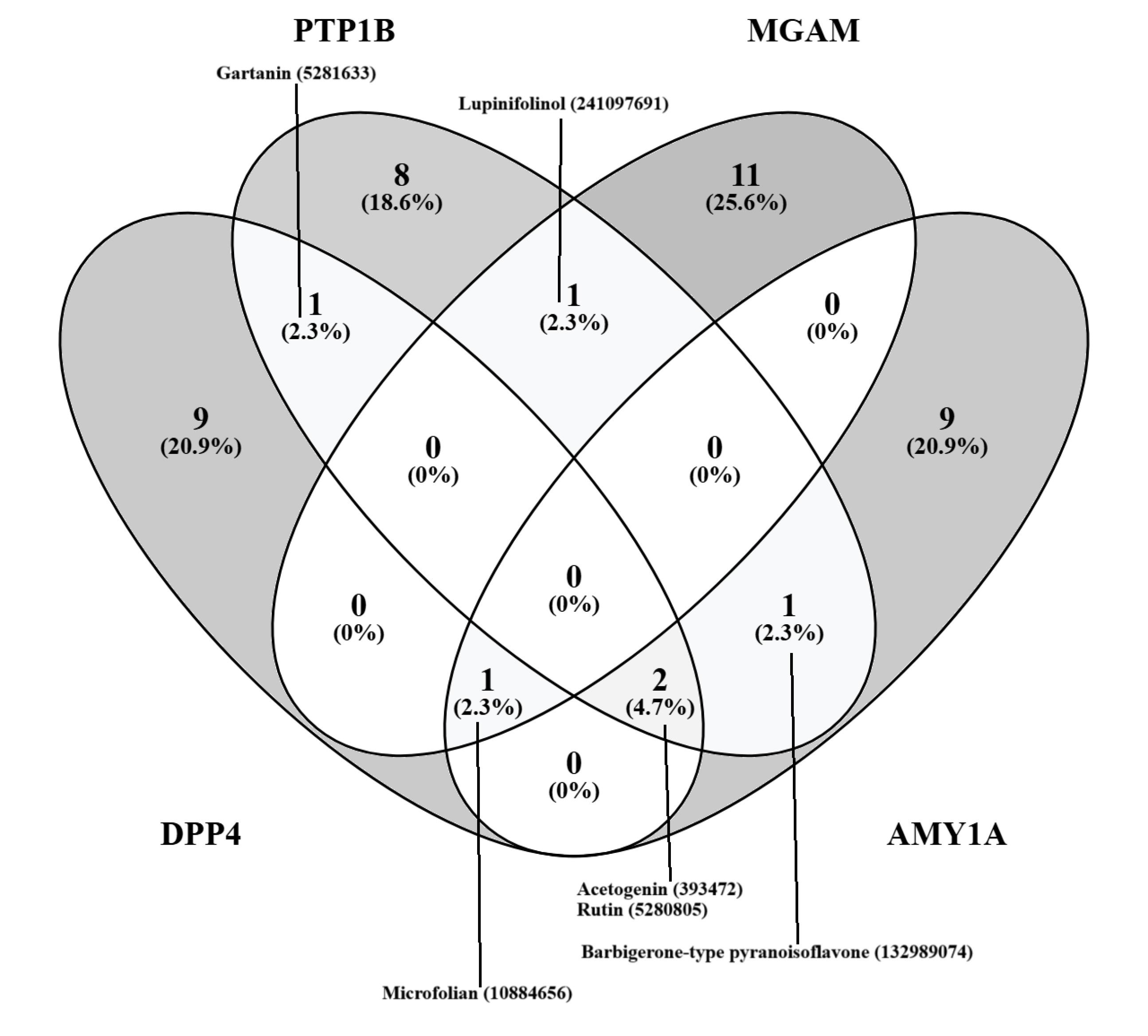
Figure 1.
Venn Diagram Showing Potentially Muti-Targeting Compounds
.
Venn Diagram Showing Potentially Muti-Targeting Compounds
Molecular Dynamics
Docking, as a drug discovery tool, has shown considerable promise, particularly in identifying the binding poses of ligands within target proteins and predicting their interactions. However, to predict a more accurate binding energy of the complexes and the stability of the simulated poses, MDs have proven to be relatively more efficient (26). This may be largely due to the fact that most docking protocols treat proteins as rigid structures and apply numerous approximations (68). Therefore, to further explore the potential of these compounds to stably interact with these targets, a molecular dynamic simulation was carried out (one compound for each target).
Molecular Mechanics Generalized Born Surface Area-Computed Binding Energies
The binding energy metric is a relatively good indicator of system stability. In protein-ligand, protein-protein, and nucleoprotein systems, noncovalent interactions significantly contribute to this stability. In the current study, the AMY1A-101679366 and DPP4-393472 complexes exhibited very low MMGBSA-computed binding energies of -65.26 kcal/mol and -54.05 kcal/mol, respectively, compared to -48.79 kcal/mol and -37.77 kcal/mol for the controls. In contrast, the MGAM-64982 and PTP1B-132988935 complexes had lower MMGBSA-computed binding energies of -21.33 kcal/mol and -19.03 kcal/mol, respectively, which were higher than those of the respective controls (Figure 2). This discrepancy may be due to the type and nature of the stabilizing non-covalent interactions. Notably, PTP1B-132988935 had fewer polar contacts relative to the control.
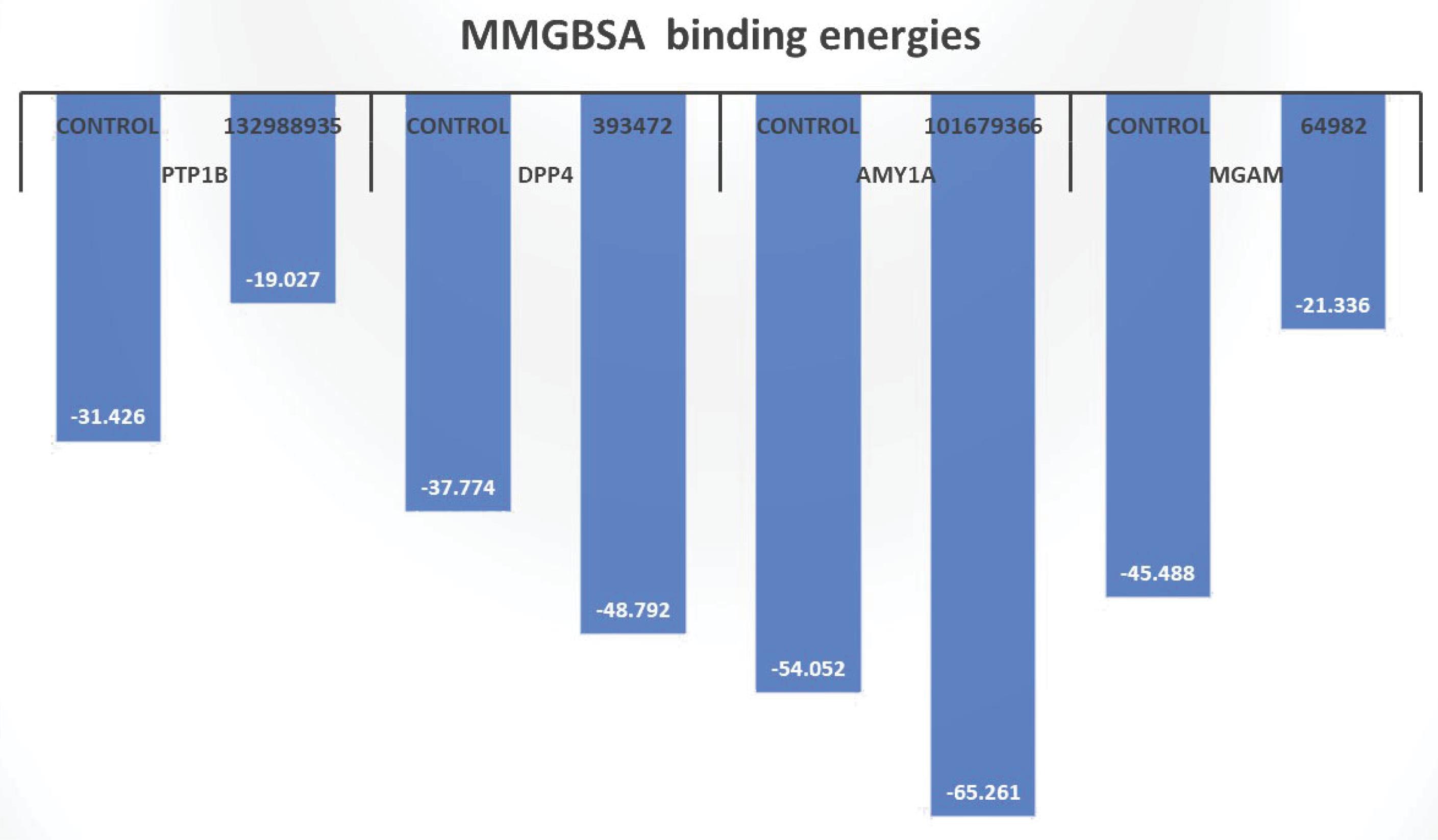
Figure 2.
MMGBSA-Computed Binding Energies for the Top-Ranked Compounds. Note. MMGBSA: Molecular Mechanics Generalized Born Surface Area
.
MMGBSA-Computed Binding Energies for the Top-Ranked Compounds. Note. MMGBSA: Molecular Mechanics Generalized Born Surface Area
Polar Contacts/Hydrogen Bond Interactions
Polar contact and hydrogen bond interactions are generally considered to be facilitators of protein-ligand binding (69). In the current study, the selected compounds showed extensive polar contacts and hydrogen bonding with important amino acid residues of their targets, comparable to the controls, except the PTP1B-132988935 complex. The interactions between compound 64982 and MGAM are seen to be stabilized by hydrogen bond interactions involving amino acid residues Asp 542, Arg 526, Asp 443, His600, and Asp 327 (Table 1, Figures S1A and S1B). This is also evident in the MD, where polar contacts and hydrogen bond interactions were observed with the residues Asp 542, Arg 526, Asp 443, His 600, and Asp 327, occurring prominently throughout the molecular dynamic simulation, as well as with residues Asp 102, Tyr 219, and Tyr 299 (Figures 3A, 3B, Figures S2A and S2B).
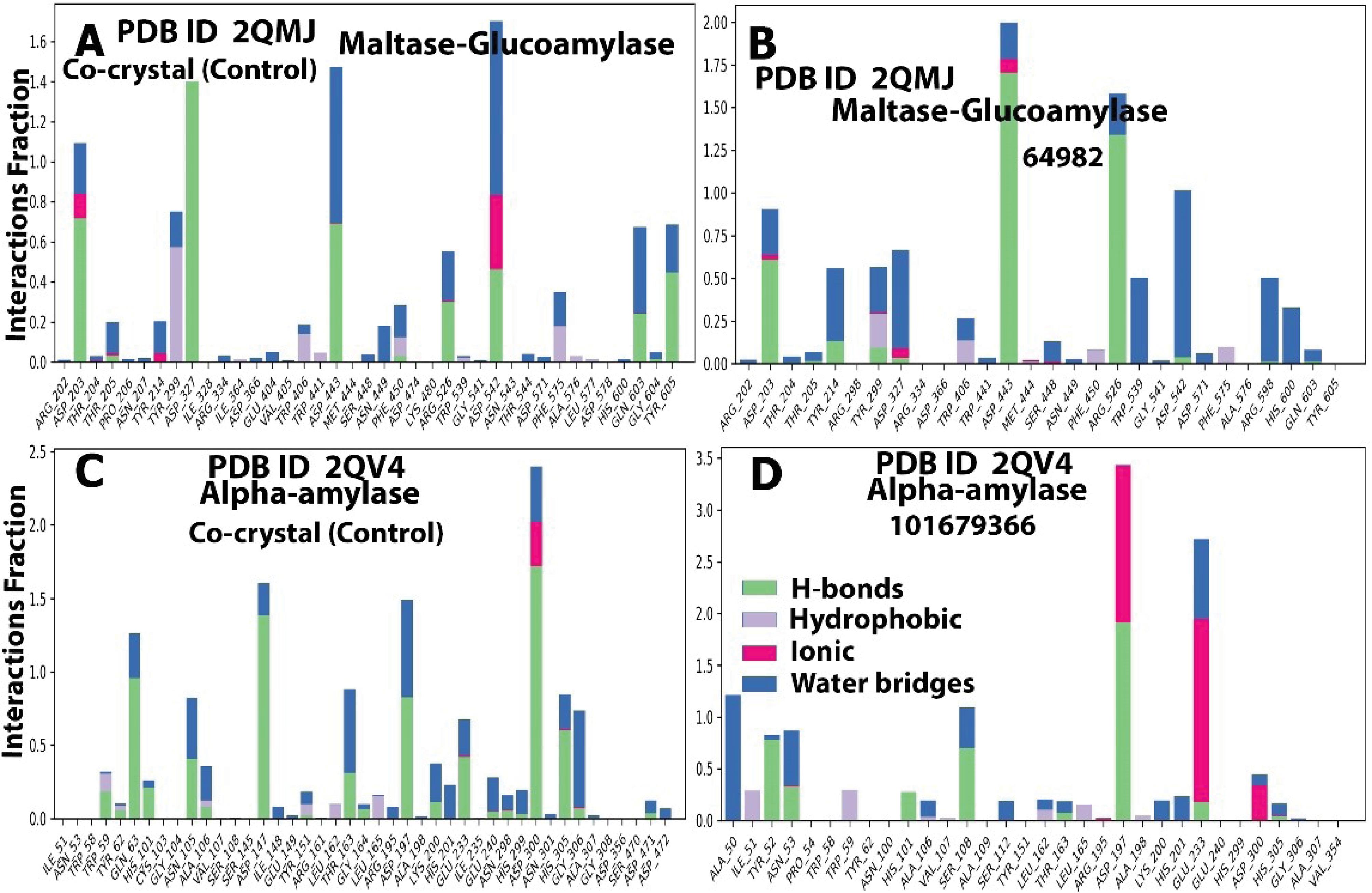
Figure 3.
Histograms Showing the Amino Acids Involved in Protein-Ligand Interactions, Types of Interactions (Polar and Non-polar), and the Proportion During the MD Trajectory for AMY1A (2QV4) and MGAM (2QMJ) Compounds and the Respective Controls. Note. MD: Molecular dynamics; AMY1A: Alpha-amylase 1; MGAM: Maltase-Glucoamylase
.
Histograms Showing the Amino Acids Involved in Protein-Ligand Interactions, Types of Interactions (Polar and Non-polar), and the Proportion During the MD Trajectory for AMY1A (2QV4) and MGAM (2QMJ) Compounds and the Respective Controls. Note. MD: Molecular dynamics; AMY1A: Alpha-amylase 1; MGAM: Maltase-Glucoamylase
In the case of the AMY1A-101679366 complex, the interactions involved hydrogen bonds with Asn 53, Ser 108, Ala 106, Asp 197, Asp 300, His 299, Glu 233, and Ala 198 (Table 1, Figures S1C and S1D), which is consistent with the proportion/number of polar contacts and hydrogen bonds observed with theses residues during the molecular dynamic simulation (Figures 3C, 3D, Figures S2C and S2D). Similarly, with compound 393472 and DPP4, the complex was stabilized through hydrogen bonding with amino acid residues Ser 209, Glu 205, Glu 206, and Tyr 670 (Table 1, Figures S1E, S1F). This is also evident from the relatively high fraction of hydrogen bonding interaction and water bridges associated with these residues during the MD simulation, as well as the high number of hydrogen bond contacts associated with the residues at every phase of the 100 ns simulation (Figures 4A, 4B, Figures S3A and S3B). These amino acid residues are considered essential in the active site of DPP4 (70) and play a significant role in stabilizing the control, a gliptin.
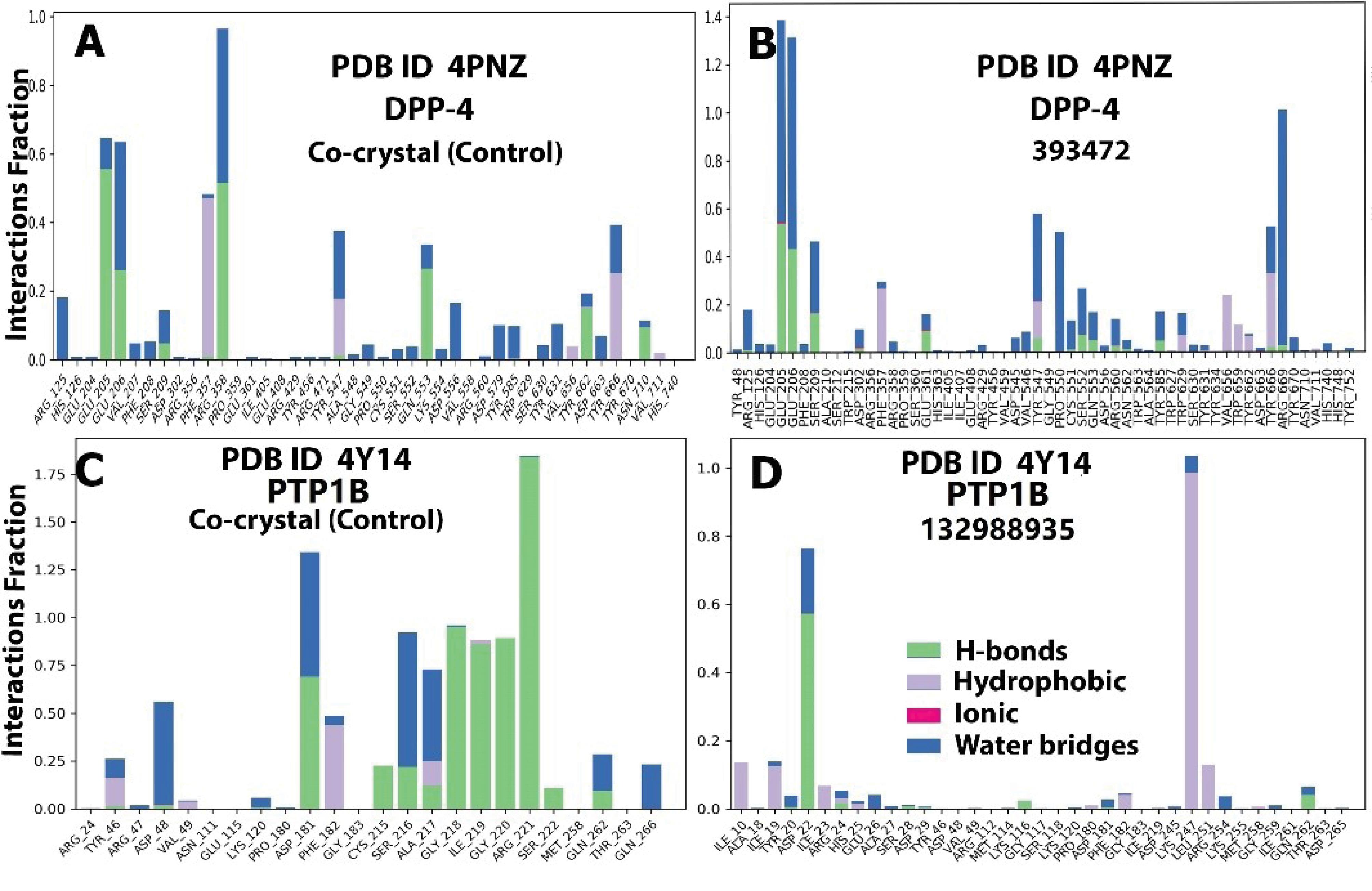
Figure 4.
Histograms Showing the Amino Acids Involved in Protein-Ligand Interactions, the Types of Interactions (Polar and Non-polar), and the Proportion During the MD Trajectory for DPP4 (4PNZ) and PTP1B (4Y14) Compounds and the Respective Controls. Note. MD: Molecular dynamics; DPP4: Dipeptidyl peptidase IV; PTP1B: Protein Tyrosine Phosphatase 1B
.
Histograms Showing the Amino Acids Involved in Protein-Ligand Interactions, the Types of Interactions (Polar and Non-polar), and the Proportion During the MD Trajectory for DPP4 (4PNZ) and PTP1B (4Y14) Compounds and the Respective Controls. Note. MD: Molecular dynamics; DPP4: Dipeptidyl peptidase IV; PTP1B: Protein Tyrosine Phosphatase 1B
For PTP1B, hydrogen bond interactions with Arg 221 and Ala 217 were observed from the post-docking interaction of compound 132988935 in PTP1B (Table 1, Figures S1G, S1H). However, the MD simulations revealed that the major polar/hydrogen bond interactions involved Asp 22, Ile 23, Ile 19, Ile 10, Lys 247, and Leu 251 (Figures 4C, 4D, Figures S3C and S3D). These interactions differed from the binding environment and interactions observed with the control. This may be attributable to conformational changes that occurred to achieve a more stable confirmation, which was slightly different from the initial docked confirmation, as mentioned earlier.
To further examine the stability of these representative complexes, MD simulations were conducted, focusing on the RMSD, RMSF, rGyr, PSA, MolSA, and SASA.
Stability of the Protein-Ligand Complexes
Root-Mean-Square Deviation Analysis
The RMSD trajectories for the MGAM, AMY1A, DPP4, and PTP1B complexes (both compound and control) are presented in Figures 5A-D and 6A-D. In the MGAM-64982 complex, the protein (Cα) displayed a fairly stable trajectory, oscillating within 0.30 Å throughout the 100 ns period, while the ligand showed an early phase fluctuation (0.5-1.8 Å), stabilizing into minor oscillations within 0.9 Å. The control exhibited a stable protein trajectory ( < 0.6 Å) after initial fluctuations around 30-40 ns (0.5-1.9 Å) and 70 ns (1.9-1.0 Å), as depicted in Figures 5A and 5B.
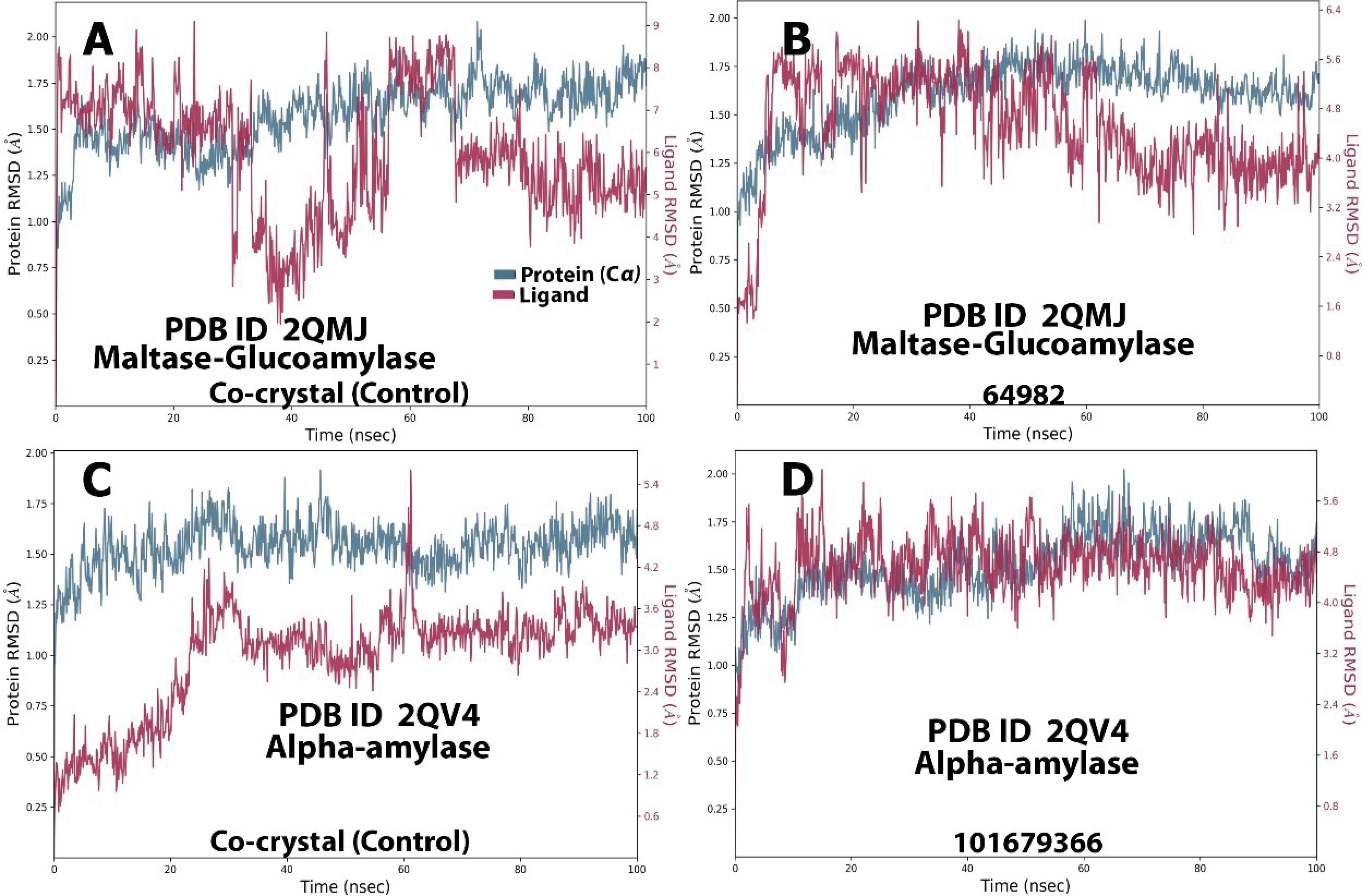
Figure 5.
Molecular Dynamics RMSD Trajectory for AMY1A (2QV4)and MGAM (2QMJ) Compounds and Their Respective Controls. Note. RMSD: Root mean square deviation; AMY1A: Alpha-amylase 1; MGAM: Maltase-Glucoamylase
.
Molecular Dynamics RMSD Trajectory for AMY1A (2QV4)and MGAM (2QMJ) Compounds and Their Respective Controls. Note. RMSD: Root mean square deviation; AMY1A: Alpha-amylase 1; MGAM: Maltase-Glucoamylase
In the AMY1A-101679366 complex, both the ligand and protein (Cα) maintained a fairly stable trajectory (1.1-1.8 Å), while the control showed a stable protein trajectory (1.30-1.40 Å) throughout the simulation. Minor fluctuations were observed for the ligand (0.25-1.4 Å) in the 0-20 ns frame, after which it remained stable ( < 2.0 Å), as illustrated in Figures 5C and 5D.
The DPP4-393472 complex exhibited a relatively stable trajectory, with protein (Cα) fluctuations around the 20 ns phase (1.2-2.5 Å), 40 ns (0.6-1.2 Å), 76 ns (0.7-2.3 Å), and 94 ns (1.5-2.4 Å). The control was also stable, except for a sharp fluctuation at 60 ns (0.25-1.75 Å), as displayed in Figures 6A and 6B.
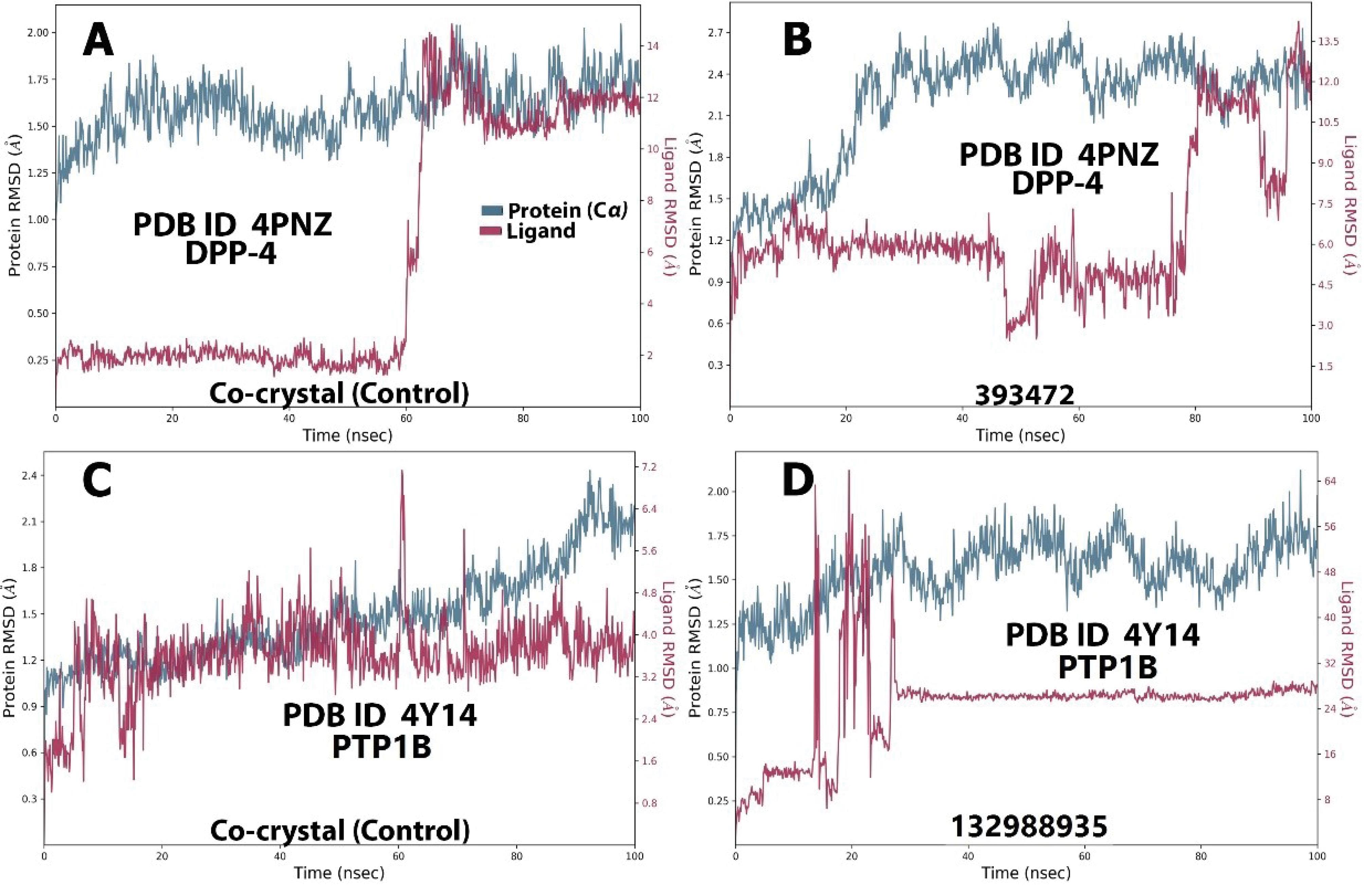
Figure 6.
Molecular Dynamics RMSD Trajectory for DPP4 (4PNZ) and PTP1B (4Y14) Compounds and Their Respective Controls. Note. RMSD: Root mean square deviation; DPP4: Dipeptidyl peptidase IV; PTP1B: Protein Tyrosine Phosphatase 1B
.
Molecular Dynamics RMSD Trajectory for DPP4 (4PNZ) and PTP1B (4Y14) Compounds and Their Respective Controls. Note. RMSD: Root mean square deviation; DPP4: Dipeptidyl peptidase IV; PTP1B: Protein Tyrosine Phosphatase 1B
The PTP1B-132988935 complex showed a stable trajectory, with the protein (Cα) oscillating within 1.05-1.75 Å for most of the simulation. The ligand, aside from fluctuations around 18-30 ns (0.25-2.00 Å), remained stable until the end of the simulation. The control displayed a stable trajectory throughout the simulation for both the protein (< 0.7 Å) and the ligand (< 0.6 Å), as depicted in Figures 6C and 6D. Overall, the RMSD for all categories remained within a fairly stable conformation for the majority of the simulations, with major fluctuations staying below 2.0 Å, indicating stable systems.
Root-Mean-Square Fluctuations Analysis
The RMSF trajectories support the RMSD data by indicating the residue numbers where major perturbations occurred (71). Consistent with the RMSD trajectory, most fluctuations remained within a stable distance of less than 2.0 Å. However, significant fluctuations were observed around a few residues, most of which coincide with important active site residues.
For the MGAM complexes (Figures S4A and S4B), the most prominent spikes were observed around residues 600 and 825 in both the control and query systems. In the AMY1A complex (Figures S4C and S4D), spikes were noted around residues 106, 108, 233, and 300. For the DPP-4 complex (Figures S5A and S5B), spikes were seen around residues 205, 206, and 250. The PTP1B complex (Figures 5C and 5D) exhibited spikes around residues 53, 181, and 215.
Radius of Gyration Analysis
The rGyr of a protein or complex is a measure of the distance of atoms from the center of mass, providing insight into the compactness of the structure (72). In this study, the MGAM-64982 complex showed rGyr fluctuations within a range of 0.9 Å (centered around 4.6 Å) compared to the control’s range of 0.9 Å (centered around 5.85 Å) (Figures S6A and S6B). The AMY1A-101679366 complex displayed rGyr oscillations within a range of 1.2 Å, centred around 8.2 Å, compared to the control’s range of 1.0 Å, centred around 5.5 Å (Figures S6C and 6D). The DPP4-393472 complex had rGyr variations between 1.7 Å (centered around 5.0 Å) compared to the control’s range of 0.6 Å (centered around 5.1 Å) (Figures S7A and S7B). For the PTP1B-132988935 complex, the rGyr varied from 0.7 Å (centered around 4.3 Å), while the control exhibited a range of 0.4 Å (centered around 3.6 Å) (Figures S7C and S7D). The results showed minor fluctuations in the trajectory ( < 2.0 Å), which did not significantly differ between the control and query complexes, except for the DPP4-393472 and PTP1B-132988935 complexes, where fluctuations differed from the controls. These differences may be due to momentary conformational changes, the sizes of the compounds, and their binding interactions with the proteins.
Polar Surface Area Analysis
PSA represents the total surface area of a molecule occupied by polar atoms and groups, providing insight into the potential for hydrogen bonding and stability (72). In the present study, the MGAM-64982 complex had a PSA ranging from 260-330 Å2 (primarily 325 Å2) compared to the control’s 500-565 Å2 (primarily 537 Å2) (Figures S6A and S6B). The AMY1A-101679366 complex had a PSA value ranging from 400-435 Å2 (primarily 420Å2) compared to the control’s 560-680 Å2 (primarily 640 Å2) (Figures S6C and S6D). The DPP4-393472 complex fluctuated within 150-240 Å2 (majorly 205 Å2), compared to the control’s 140-170 Å2 (primarily 152 Å2) (Figures S7A and S7B). The PTP1B-132988935 complex ranged from 85-120 Å2 (majorly 90 Å2) compared to the control’s 215-255 Å2 (primarily 240 Å2) (Figures S7C and S7D). Higher PSA values were observed for AMY1A-101679366, MGAM-64982, and DPP4-393472 complexes, reflecting the higher number of polar contacts/ hydrogen bond interactions observed, as well as their stability.
Molecular Surface Area Analysis
MolSA refers to the total surface area of the ligand in contact with its environment. The MGAM-64982 complex had MolSA values ranging from 490-510 Å2 (primarily 505 Å2) compared to the control’s 360-407 Å2 (primarily 360 Å2) (Figures S6A and S6B). The AMY1A-101679366 complex had MolSA values ranging from 550-610 Å2 (primarily 600 Å2) compared to the control’s 525-540 Å2 (primarily 530 Å2) (Figures S6C and S6D). The DPP4-393472 complex fluctuated within 352-365 Å2 (primarily 358 Å2), compared to the control’s 460-500 Å2 (primarily 485 Å2) (Figures S7A and S7B). The PTP1B-132988935 complex ranged from 328-344 Å2 (primarily 336 Å2) compared to the control’s 310-380 Å2 (primarily 345 Å2) (Figures S7C and S7D).
Solvent Accessible Surface Area Analysis
SASA represents the portion of the ligand’s surface accessible to solvent molecules. The MGAM-64982 complex had SASA values ranging from 160-320 Å2 (primarily 200 Å2) compared to the control’s 240-480 Å2 (primarily 320 Å2) (Figures S6A and S6B). The AMY1A-101679366 complex had SASA values ranging from 210-380 Å2 (primarily 300 Å2) compared to the control’s 205-400 Å2 (primarily 320 Å2) (Figures S6C and S6D). The DPP4-393472 complex fluctuated within 150-450 Å2 (majorly 200 Å2), compared to the control’s 150-470 Å2 (majorly 310 Å2) (Figures S7A and S6B). The PTP1B-132988935 complex ranged from 200-800 Å2 (primarily 180 Å2) compared to the control’s 150-300 Å2 (primarily 240 Å2) (Figures S7C and S7D). Higher SASA values were observed for the PTP1B-132988935 complex, indicating greater solvent exposure, which could contribute to its reduced stability. MGAM-64982, AMY1A-101679366, and DPP4-393472 complexes had SASA values comparable to their controls, indicating similar solvent exposure.
Conclusion
In conclusion, this study employed virtual screening techniques, including molecular docking and MD simulations, to identify potentially active antidiabetic compounds from African medicinal plants. Several promising compounds were identified, including basic flavonoids, their glycosides, prenylated and acetylated derivatives, xanthones, alkaloids, iridoid/secoiridoid glycosides, and terpenoids, which exhibited stable binding interactions in simulations. Based on these findings, future research should prioritize these compounds for in vitro and in vivo assays to confirm their bioactivity and pharmacokinetics. Additionally, these findings not only prioritize compounds for in vitro and in vivo assays to confirm their bioactivity but also suggest specific molecular scaffolds with potential for optimization. For drug development, these identified compounds offer promising starting points for lead compound development, with potential practical applications in designing novel antidiabetic drugs that are potentially safer, more accessible, and cost-effective than current options.
Study Limitations
The computational approach that relied on molecular docking and dynamics simulations may not fully capture the in vivo complexity of target proteins, including their flexibility, water-mediated interactions, and allosteric effects. Additionally, the scoring functions used in simulations can introduce inaccuracies in ranking compounds and may lead to false positives. Moreover, due to the computational intensity and high-power demands of MD simulations, the study could not perform MD for all high-ranking compounds or conduct multiple simulations for selected compounds. Experimental validation is necessary to confirm these predictions.
Authors’ Contribution
Conceptualization: Chimaobi James Ononamadu.
Data curation: Chimaobi James Ononamadu, Godwin Okwudiri Ihegboro.
Formal analysis: Chimaobi James Ononamadu, Mohnad Abdalla.
Investigation: Chimaobi James Ononamadu, Mohnad Abdalla.
Methodology: Chimaobi James Ononamadu.
Writing-original draft: Chimaobi James Ononamadu.
Writing-review & editing: Chimaobi James Ononamadu, Godwin Okwudiri Ihegboro.
Competing Interests
All authors declare that they have no conflict of interests.
Ethical Approval
Not Applicable
Funding
This work was supported by the Tertiary Education Trust Fund (TETFUND) IBR Grant [TETF/DR&D/UNI/WUDIL/RG/2018/VOL1.].
Supplementary Files
Supplementary file 1 contains Tables S1-S5 and Figures S1-S7.
(pdf)
References
- Aynalem SB, Zeleke AJ. Prevalence of diabetes mellitus and its risk factors among individuals aged 15 years and above in Mizan-Aman town, southwest Ethiopia, 2016: a cross sectional study. Int J Endocrinol 2018; 2018:9317987. doi: 10.1155/2018/9317987 [Crossref] [ Google Scholar]
- Ibrahim MT, Uzairu A, Shallangwa GA, Ibrahim A. In-silico studies of some oxadiazoles derivatives as anti-diabetic compounds. J King Saud Univ Sci 2020; 32(1):423-32. doi: 10.1016/j.jksus.2018.06.006 [Crossref] [ Google Scholar]
- Dilworth L, Facey A, Omoruyi F. Diabetes mellitus and its metabolic complications: the role of adipose tissues. Int J Mol Sci 2021; 22(14):7644. doi: 10.3390/ijms22147644 [Crossref] [ Google Scholar]
- Raveendran AV, Chacko EC, Pappachan JM. Non-pharmacological treatment options in the management of diabetes mellitus. Eur Endocrinol 2018; 14(2):31-9. doi: 10.17925/ee.2018.14.2.31 [Crossref] [ Google Scholar]
- 5Antar SA, Ashour NA, Sharaky M, Khattab M, Ashour NA, Zaid RT. Diabetes mellitus: classification, mediators, and complications; a gate to identify potential targets for the development of new effective treatments. Biomed Pharmacother 2023; 168:115734. doi: 10.1016/j.biopha.2023.115734 [Crossref] [ Google Scholar]
- Ogbera AO, Ekpebegh C. Diabetes mellitus in Nigeria: the past, present and future. World J Diabetes 2014; 5(6):905-11. doi: 10.4239/wjd.v5.i6.905 [Crossref] [ Google Scholar]
- Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract 2022; 183:109119. doi: 10.1016/j.diabres.2021.109119 [Crossref] [ Google Scholar]
- Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract 2019; 157:107843. doi: 10.1016/j.diabres.2019.107843 [Crossref] [ Google Scholar]
- Alam S, Hasan MK, Neaz S, Hussain N, Hossain MF, Rahman T. Diabetes mellitus: insights from epidemiology, biochemistry, risk factors, diagnosis, complications and comprehensive management. Diabetology 2021; 2(2):36-50. doi: 10.3390/diabetology2020004 [Crossref] [ Google Scholar]
- Chaudhury A, Duvoor C, Reddy Dendi VS, Kraleti S, Chada A, Ravilla R. Clinical review of antidiabetic drugs: implications for type 2 diabetes mellitus management. Front Endocrinol (Lausanne) 2017; 8:6. doi: 10.3389/fendo.2017.00006 [Crossref] [ Google Scholar]
- Sofowora A, Ogunbodede E, Onayade A. The role and place of medicinal plants in the strategies for disease prevention. Afr J Tradit Complement Altern Med 2013; 10(5):210-29. doi: 10.4314/ajtcam.v10i5.2 [Crossref] [ Google Scholar]
- Farzaei F, Morovati MR, Farjadmand F, Farzaei MH. A mechanistic review on medicinal plants used for diabetes mellitus in traditional Persian medicine. J Evid Based Complementary Altern Med 2017; 22(4):944-55. doi: 10.1177/2156587216686461 [Crossref] [ Google Scholar]
- Furman BL, Candasamy M, Bhattamisra SK, Veettil SK. Reduction of blood glucose by plant extracts and their use in the treatment of diabetes mellitus; discrepancies in effectiveness between animal and human studies. J Ethnopharmacol 2020; 247:112264. doi: 10.1016/j.jep.2019.112264 [Crossref] [ Google Scholar]
- Ntie-Kang F, Lifongo LL, Mbah JA, Owono Owono LC, Megnassan E, Mbaze LM. In silico drug metabolism and pharmacokinetic profiles of natural products from medicinal plants in the Congo basin. In Silico Pharmacol 2013; 1:12. doi: 10.1186/2193-9616-1-12 [Crossref] [ Google Scholar]
- Riyaphan J, Jhong CH, Lin SR, Chang CH, Tsai MJ, Lee DN. Hypoglycemic efficacy of docking selected natural compounds against α-glucosidase and α-amylase. Molecules 2018; 23(9):2260. doi: 10.3390/molecules23092260 [Crossref] [ Google Scholar]
- Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 2011; 7(2):146-57. doi: 10.2174/157340911795677602 [Crossref] [ Google Scholar]
- Bibi S, Kulsoom S, Rashid H. In silico approach for lead identification and optimization of antidiabetic compounds. IOSR J Pharm Biol Sci 2013; 7(3):36-46. [ Google Scholar]
- Egan WJ, Merz KM Jr, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem 2000; 43(21):3867-77. doi: 10.1021/jm000292e [Crossref] [ Google Scholar]
- Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery 1 A qualitative and quantitative characterization of known drug databases. J Comb Chem 1999; 1(1):55-68. doi: 10.1021/cc9800071 [Crossref] [ Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001; 46(1-3):3-26. doi: 10.1016/s0169-409x(00)00129-0 [Crossref] [ Google Scholar]
- Muegge I, Heald SL, Brittelli D. Simple selection criteria for drug-like chemical matter. J Med Chem 2001; 44(12):1841-6. doi: 10.1021/jm015507e [Crossref] [ Google Scholar]
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002; 45(12):2615-23. doi: 10.1021/jm020017n [Crossref] [ Google Scholar]
- Ganesan A. The impact of natural products upon modern drug discovery. Curr Opin Chem Biol 2008; 12(3):306-17. doi: 10.1016/j.cbpa.2008.03.016 [Crossref] [ Google Scholar]
- Macarrón R, Luengo JI. Yin and Yang in medicinal chemistry: what does drug-likeness mean?. Future Med Chem 2011; 3(5):505-7. doi: 10.4155/fmc.11.19 [Crossref] [ Google Scholar]
- Zhang MQ, Wilkinson B. Drug discovery beyond the ‘rule-of-five’. Curr Opin Biotechnol 2007; 18(6):478-88. doi: 10.1016/j.copbio.2007.10.005 [Crossref] [ Google Scholar]
- Ononamadu CJ, Abdalla M, Ihegboro GO, Li J, Owolarafe TA, John TD. In silico identification and study of potential anti-mosquito juvenile hormone binding protein (MJHBP) compounds as candidates for dengue virus - vector insecticides. Biochem Biophys Rep 2021; 28:101178. doi: 10.1016/j.bbrep.2021.101178 [Crossref] [ Google Scholar]
- Ramírez D, Caballero J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data?. Molecules 2018; 23(5):1038. doi: 10.3390/molecules23051038 [Crossref] [ Google Scholar]
- Galli CL, Sensi C, Fumagalli A, Parravicini C, Marinovich M, Eberini I. A computational approach to evaluate the androgenic affinity of iprodione, procymidone, vinclozolin and their metabolites. PLoS One 2014; 9(8):e104822. doi: 10.1371/journal.pone.0104822 [Crossref] [ Google Scholar]
- Nissink JW. Simple size-independent measure of ligand efficiency. J Chem Inf Model 2009; 49(6):1617-22. doi: 10.1021/ci900094m [Crossref] [ Google Scholar]
- Baby K, Maity S, Mehta CH, Suresh A, Nayak UY, Nayak Y. Targeting SARS-CoV-2 RNA-dependent RNA polymerase: an in silico drug repurposing for COVID-19. F1000Res 2020; 9:1166. doi: 10.12688/f1000research.26359.1 [Crossref] [ Google Scholar]
- Jin Z, Wang Y, Yu XF, Tan QQ, Liang SS, Li T. Structure-based virtual screening of influenza virus RNA polymerase inhibitors from natural compounds: molecular dynamics simulation and MM-GBSA calculation. Comput Biol Chem 2020; 85:107241. doi: 10.1016/j.compbiolchem.2020.107241 [Crossref] [ Google Scholar]
- Mohammed A, Ibrahim MA, Islam MS. African medicinal plants with antidiabetic potentials: a review. Planta Med 2014; 80(5):354-77. doi: 10.1055/s-0033-1360335 [Crossref] [ Google Scholar]
- Mohammed A. Hypoglycemic potential of African medicinal plants in diabetic and non-diabetic human subjects: a review. Clin Complement Med Pharmacol 2023; 3(2):100081. doi: 10.1016/j.ccmp.2023.100081 [Crossref] [ Google Scholar]
- van de Venter M, Roux S, Bungu LC, Louw J, Crouch NR, Grace OM. Antidiabetic screening and scoring of 11 plants traditionally used in South Africa. J Ethnopharmacol 2008; 119(1):81-6. doi: 10.1016/j.jep.2008.05.031 [Crossref] [ Google Scholar]
- Pereira AS, den Haan H, Peña-García J, Moreno MM, Pérez-Sánchez H, Apostolides Z. Exploring African medicinal plants for potential anti-diabetic compounds with the DIA-DB inverse virtual screening web server. Molecules 2019; 24(10):2002. doi: 10.3390/molecules24102002 [Crossref] [ Google Scholar]
- Dandekar PD, Kotmale AS, Chavan SR, Kadlag PP, Sawant SV, Dhavale DD. Insights into the inhibition mechanism of human pancreatic α-amylase, a type 2 diabetes target, by dehydrodieugenol B isolated from Ocimumtenuiflorum. ACS Omega 2021; 6(3):1780-6. doi: 10.1021/acsomega.0c00617 [Crossref] [ Google Scholar]
- Kaur N, Kumar V, Nayak SK, Wadhwa P, Kaur P, Sahu SK. Alpha-amylase as molecular target for treatment of diabetes mellitus: a comprehensive review. Chem Biol Drug Des 2021; 98(4):539-60. doi: 10.1111/cbdd.13909 [Crossref] [ Google Scholar]
- Eleftheriou P, Geronikaki A, Petrou A. PTP1b inhibition, a promising approach for the treatment of diabetes type II. Curr Top Med Chem 2019; 19(4):246-63. doi: 10.2174/1568026619666190201152153 [Crossref] [ Google Scholar]
- Röhrborn D, Wronkowitz N, Eckel J. DPP4 in diabetes. Front Immunol 2015; 6:386. doi: 10.3389/fimmu.2015.00386 [Crossref] [ Google Scholar]
- Saini K, Sharma S, Khan Y. DPP-4 inhibitors for treating T2DM - hype or hope? an analysis based on the current literature. Front Mol Biosci 2023; 10:1130625. doi: 10.3389/fmolb.2023.1130625 [Crossref] [ Google Scholar]
- Nguyen Vo TH, Tran N, Nguyen D, Le L. An in silico study on antidiabetic activity of bioactive compounds in Euphorbia thymifolia Linn. Springerplus 2016; 5(1):1359. doi: 10.1186/s40064-016-2631-5 [Crossref] [ Google Scholar]
- Damián-Medina K, Salinas-Moreno Y, Milenkovic D, Figueroa-Yáñez L, Marino-Marmolejo E, Higuera-Ciapara I. In silico analysis of antidiabetic potential of phenolic compounds from blue corn (Zea mays L) and black bean (Phaseolus vulgaris L). Heliyon 2020; 6(3):e03632. doi: 10.1016/j.heliyon.2020.e03632 [Crossref] [ Google Scholar]
- Macalalad MA, Gonzales AA 3rd. In silico screening and identification of antidiabetic inhibitors sourced from phytochemicals of Philippine plants against four protein targets of diabetes (PTP1B, DPP-4, SGLT-2, and FBPase). Molecules 2023; 28(14):5301. doi: 10.3390/molecules28145301 [Crossref] [ Google Scholar]
- Al-Abbasi FA. Potential antidiabetic activity of barbigerone on glucose and inflammatory cytokine levels in streptozotocin activated diabetic rats. J King Saud Univ Sci 2022; 34(7):102249. doi: 10.1016/j.jksus.2022.102249 [Crossref] [ Google Scholar]
- Hernández Navia SE, Figueroa-Hernández JL, Rodríguez-Zavala JS, Rodriguez-Sosa M, Martínez-Vázquez M. Anti-diabetic effects of cucurbitacins from Ibervillealindheimeri on induced mouse diabetes. J Chem 2022; 2022(1):3379557. doi: 10.1155/2022/3379557 [Crossref] [ Google Scholar]
- Sang J, Dhakal S, Lee Y. Cucurbitacin B suppresses hyperglycemia associated with a high sugar diet and promotes sleep in Drosophila melanogaster. Mol Cells 2021; 44(2):68-78. doi: 10.14348/molcells.2021.2245 [Crossref] [ Google Scholar]
- Safaei R, Sakhaee K, Saberifar M, Fadaei MS, EdalatJoo S, Fadaei MR. Mechanistic insights into the xanthones present in mangosteen fruit (Garcinia mangostana) and their applications in diabetes and related complications. J Food Biochem 2023; 2023(1):5334312. doi: 10.1155/2023/5334312 [Crossref] [ Google Scholar]
- Zengin G, El-Raey M, El-Kashak W, Batiha GE, Althumairy D, Alamer S. Sweroside: an iridoid glycoside of potential neuroprotective, antidiabetic, and antioxidant activities supported by molecular docking. Amino Acids 2023; 55(12):1765-74. doi: 10.1007/s00726-023-03262-9 [Crossref] [ Google Scholar]
- Kumar S, Niguram P, Jairaj V, Chauhan N, Jinagal S, Sagar S, et al. Exploring the potential of semi-synthetic Swertiamarin analogues for GLUT facilitation and insulin secretion in NIT-1 cell lines: a molecular docking and in-vitro study. Nat Prod Res. 2024:1-5. 10.1080/14786419.2024.2342005.
- Trinh DH, Tran PT, Trinh BT, Nguyen HT, Nguyen HD, Ha LD. Coumarins and acridone alkaloids with α-glucosidase inhibitory and antioxidant activity from the roots of Paramignyatrimera. Phytochemistry Letters 2020; 35:94-8. doi: 10.1016/j.phytol.2019.10.010 [Crossref] [ Google Scholar]
- Behl T, Gupta A, Albratty M, Najmi A, Meraya AM, Alhazmi HA. Alkaloidal phytoconstituents for diabetes management: exploring the unrevealed potential. Molecules 2022; 27(18):5851. doi: 10.3390/molecules27185851 [Crossref] [ Google Scholar]
- Muhammad I, Rahman N, Nishan U, Shah M. Antidiabetic activities of alkaloids isolated from medicinal plants. Braz J Pharm Sci 2021; 57:e19130. doi: 10.1590/s2175-97902020000419130 [Crossref] [ Google Scholar]
- Pulbutr P, Nantana P, Suksabai S, Mudjupa C, Denchai R, Rattanakiat S. Inhibitory actions of lupinifolin isolated from Derris reticulata stem against carbohydrate-digesting enzymes. Pharmacogn Res 2020; 12(2):102-6. doi: 10.4103/pr.pr_117_19 [Crossref] [ Google Scholar]
- Bazyar H, Moradi L, Zaman F, Zare Javid A. The effects of rutin flavonoid supplement on glycemic status, lipid profile, atherogenic index of plasma, brain-derived neurotrophic factor (BDNF), some serum inflammatory, and oxidative stress factors in patients with type 2 diabetes mellitus: a double-blind, placebo-controlled trial. Phytother Res 2023; 37(1):271-84. doi: 10.1002/ptr.7611 [Crossref] [ Google Scholar]
- Ghorbani A. Mechanisms of antidiabetic effects of flavonoid rutin. Biomed Pharmacother 2017; 96:305-12. doi: 10.1016/j.biopha.2017.10.001 [Crossref] [ Google Scholar]
- Mathrani A, Yip W, Sequeira-Bisson IR, Barnett D, Stevenson O, Taylor MW. Effect of a 12-week polyphenol rutin intervention on markers of pancreatic β-cell function and gut microbiota in adults with overweight without diabetes. Nutrients 2023; 15(15):3360. doi: 10.3390/nu15153360 [Crossref] [ Google Scholar]
- Shi GJ, Li Y, Cao QH, Wu HX, Tang XY, Gao XH. In vitro and in vivo evidence that quercetin protects against diabetes and its complications: a systematic review of the literature. Biomed Pharmacother 2019; 109:1085-99. doi: 10.1016/j.biopha.2018.10.130 [Crossref] [ Google Scholar]
- Alaaeldin R, Abdel-Rahman IA, Hassan HA, Youssef N, Allam AE, Abdelwahab SF. Carpachromene ameliorates insulin resistance in HepG2 cells via modulating IR/IRS1/PI3k/Akt/GSK3/FoxO1 pathway. Molecules 2021; 26(24):7629. doi: 10.3390/molecules26247629 [Crossref] [ Google Scholar]
- Li ZP, Song YH, Uddin Z, Wang Y, Park KH. Inhibition of protein tyrosine phosphatase 1B (PTP1B) and α-glucosidase by xanthones from Cratoxylumcochinchinense, and their kinetic characterization. Bioorg Med Chem 2018; 26(3):737-46. doi: 10.1016/j.bmc.2017.12.043 [Crossref] [ Google Scholar]
- Cardozo-Muñoz J, Cuca-Suárez LE, Prieto-Rodríguez JA, Lopez-Vallejo F, Patiño-Ladino OJ. Multitarget action of xanthones from Garcinia mangostana against α-amylase, α-glucosidase and pancreatic lipase. Molecules 2022; 27(10):3283. doi: 10.3390/molecules27103283 [Crossref] [ Google Scholar]
- Singh AK, Patel PK, Choudhary K, Joshi J, Yadav D, Jin JO. Quercetin and coumarin inhibit dipeptidyl peptidase-IV and exhibits antioxidant properties: in silico, in vitro, ex vivo. Biomolecules 2020; 10(2):207. doi: 10.3390/biom10020207 [Crossref] [ Google Scholar]
- Choi HJ, Bae EY, No YJ, Baek SH. Protein tyrosine phosphatase 1B activity of quercetin from Houttuynia cordata. J Physiol Pathol Korean Med 2008; 22(6):1532-6. [ Google Scholar]
- Oboh G, Ademosun AO, Ayeni PO, Omojokun OS, Bello F. Comparative effect of quercetin and rutin on α-amylase, α-glucosidase, and some pro-oxidant-induced lipid peroxidation in rat pancreas. Comp Clin Path 2015; 24(5):1103-10. doi: 10.1007/s00580-014-2040-5 [Crossref] [ Google Scholar]
- Jang J, Na M, Thuong PT, Njamen D, Mbafor JT, Fomum ZT. Prenylated flavonoids with PTP1B inhibitory activity from the root bark of Erythrina mildbraedii. Chem Pharm Bull (Tokyo) 2008; 56(1):85-8. doi: 10.1248/cpb.56.85 [Crossref] [ Google Scholar]
- Salinas-Arellano E, Pérez-Vásquez A, Rivero-Cruz I, Torres-Colin R, González-Andrade M, Rangel-Grimaldo M. Flavonoids and terpenoids with PTP-1B inhibitory properties from the infusion of Salvia amarissima Ortega. Molecules 2020; 25(15):3530. doi: 10.3390/molecules25153530 [Crossref] [ Google Scholar]
- Agrawal D, Chourasia A, Ganeshpurkar A, Shrivastava A, Dubey N. In vitro α-amylase and α-glucosidase inhibitory potential of Pleurotusostreatus cv Florida extract. Indian J Biochem Biophys 2022; 59(10):1016-9. doi: 10.56042/ijbb.v59i10.29484 [Crossref] [ Google Scholar]
- Suthiphasilp V, Rujanapun N, Kumboonma P, Chaiyosang B, Tontapha S, Maneerat T. Antidiabetic and cytotoxic activities of rotenoids and isoflavonoids isolated from Millettiapachycarpa Benth. ACS Omega 2022; 7(28):24511-21. doi: 10.1021/acsomega.2c02163 [Crossref] [ Google Scholar]
- Radwan A, Mahrous GM. Docking studies and molecular dynamics simulations of the binding characteristics of waldiomycin and its methyl ester analog to Staphylococcus aureus histidine kinase. PLoS One 2020; 15(6):e0234215. doi: 10.1371/journal.pone.0234215 [Crossref] [ Google Scholar]
- Chen D, Oezguen N, Urvil P, Ferguson C, Dann SM, Savidge TC. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci Adv 2016; 2(3):e1501240. doi: 10.1126/sciadv.1501240 [Crossref] [ Google Scholar]
- Pantaleão SQ, Philot EA, de Resende-Lara PT, Lima AN, Perahia D, Miteva MA. Structural dynamics of DPP-4 and its influence on the projection of bioactive ligands. Molecules 2018; 23(2):490. doi: 10.3390/molecules23020490 [Crossref] [ Google Scholar]
- Shahraki O, Zargari F, Edraki N, Khoshneviszadeh M, Firuzi O, Miri R. Molecular dynamics simulation and molecular docking studies of 1,4-dihydropyridines as P-glycoprotein’s allosteric inhibitors. J Biomol Struct Dyn 2018; 36(1):112-25. doi: 10.1080/07391102.2016.1268976 [Crossref] [ Google Scholar]
- Bagewadi ZK, Yunus Khan TM, Gangadharappa B, Kamalapurkar A, Mohamed Shamsudeen S, Yaraguppi DA. Molecular dynamics and simulation analysis against superoxide dismutase (SOD) target of Micrococcus luteus with secondary metabolites from Bacillus licheniformis recognized by genome mining approach. Saudi J Biol Sci 2023; 30(9):103753. doi: 10.1016/j.sjbs.2023.103753 [Crossref] [ Google Scholar]