Avicenna Journal of Medical Biochemistry. 12(2):123-130.
doi: 10.34172/ajmb.2567
Original Article
Natural Anthraquinones as Potential Akt1-Targeted Anticancer Agents
Shokoofeh Jamshidi 1  , Fatemeh Mahfouzi 1 , Setareh Shojaei 1 , Amir Taherkhani 2, *
, Fatemeh Mahfouzi 1 , Setareh Shojaei 1 , Amir Taherkhani 2, *
Author information:
1Department of Oral and Maxillofacial Pathology, School of Dentistry, Hamadan University of Medical Sciences, Hamadan, Iran
2Research Center for Molecular Medicine, Institute of Cancer, Avicenna Health Research Institute, Hamadan University of Medical Sciences, Hamadan, Iran
Abstract
Background: The phosphoinositide 3-kinase/protein kinase B (Akt)/mammalian target of the rapamycin signaling pathway is crucial in cancer progression. Akt1, a vital pathway component, has emerged as a promising therapeutic target.
Objectives: This study used molecular docking analysis to investigate the potential of anthraquinones (AQs) as Akt1 inhibitors.
Methods: The crystallographic structure of Akt1 was obtained from the Protein Data Bank (PDB ID: 4GV1). Twenty-one AQ compounds were selected for docking analyses using AutoDock 4.0. Binding affinities and interaction modes were compared with two Akt1 reference inhibitors.
Results: Eleven AQs demonstrated substantial binding affinity to the Akt1’s catalytic site at nanomolar concentrations. Hypericin and sennidin B exhibited the most potent inhibitory effects, with ΔGbinding values of -11.19 kcal/mol and -10.36 kcal /mol, respectively, surpassing control inhibitors. Hypericin formed three hydrogen bonds and two hydrophobic interactions with the Akt1 catalytic cleft, while sennidin B formed six hydrogen and one hydrophobic interaction.
Conclusion: This study identified several AQs, particularly hypericin and sennidin B, as promising Akt1 inhibitors with superior binding affinities compared to reference compounds. These findings provide a foundation for further developing AQ-based Akt1-targeted therapeutics in cancer treatment. Future research should focus on the in vitro and in vivo validation of these compounds’ efficacy and safety profiles.
Keywords: Akt1, Anthraquinone, Cancer, Drug, Molecular docking, Traditional medicine,
Copyright and License Information
© 2024 The Author(s); Published by Hamadan University of Medical Sciences.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium provided the original work is properly cited.
Please cite this article as follows: Jamshidi S, Mahfouzi F, Shojaei S, Taherkhani A. Natural anthraquinones as potential Akt1-targeted anticancer agents. Avicenna J Med Biochem. 2024; 12(2):123-130. doi:10.34172/ajmb.2567
Background
Cancer remains a leading cause of death globally, with nearly 2 million new cases projected in the United States alone for 2023 (1). The phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) signaling pathway is central to cancer initiation and progression across diverse malignancies. Its influence extends from metabolic reprogramming to drug resistance mechanisms, making it a prime target for therapeutic interventions. In bladder cancer, the hyperactivation of the PI3K/Akt/mTOR pathway significantly contributes to the Warburg effect, a metabolic shift favoring aerobic glycolysis (2). Furthermore, the deregulation of protein synthesis via the PI3K/Akt/mTOR pathway has emerged as a critical driver of colorectal cancer development. Clinical trials using mTOR inhibitors and other targeted therapies have shown promise in overcoming resistance mechanisms in colorectal cancer (3).
Interestingly, metformin, a drug used for diabetes management, has exhibited anticancer properties. By inhibiting the PI3K/Akt/mTOR pathway, metformin enhanced the effectiveness of standard chemotherapeutics in ovarian cancer (4). The PI3K/Akt/mTOR signaling pathway is crucial in developing and progressing oral cancer, particularly oral squamous cell carcinoma (OSCC). It influences cellular processes, including proliferation, survival, angiogenesis, and metastasis (5). Multiple studies have explored the role of this pathway in OSCC through various molecular mechanisms and therapeutic interventions. In this regard, Su et al (6) focused on radiosensitization strategies for head and neck squamous cell carcinomas, highlighting that targeting the PI3K/Akt/mTOR pathway pharmacologically can improve radiotherapy outcomes by overcoming radioresistance.
Within the PI3K/AKT/mTOR pathway, Akt1 acts as a critical control point. Consequently, inhibiting Akt1 has emerged as a promising therapeutic strategy in cancer treatment. Costunolide represented a dual inhibitory effect on MEK1 and Akt1/2, confirming efficacy in overcoming resistance to osimertinib, an EGFR-tyrosine kinase inhibitor in lung cancer (7). Further emphasizing the clinical value of targeting Akt1 mutations, Smyth et al (8) investigated capivasertib, an Akt kinase inhibitor. Their findings demonstrated promising activity in heavily pretreated patients with estrogen receptor-positive metastatic breast cancer harboring Akt1 E17K mutations, either as monotherapy or combined with fulvestrant. These studies suggest that targeting Akt1 holds promise for cancer treatment, particularly in overcoming resistance to existing therapies.
The relentless quest for safer and more productive cancer therapies has ignited research interest in natural products, especially those derived from plants. This burgeoning fascination is rooted in the potential of these natural compounds to offer fewer adverse effects than conventional chemotherapeutic agents (9). Anthraquinones (AQs), a class of polycyclic compounds ubiquitous in numerous plant species, have emerged as promising contenders in this exploration. These naturally occurring pigments boast diverse biological activities, including anticancer, antibacterial, and antioxidant properties, which may contribute to mitigating disease risk (10). Notably, the core structural motif of AQs forms the backbone of several well-established clinical anticancer drugs. However, the ever-looming specter of drug resistance in cancer underscores the pressing need for the development of novel therapeutic modalities. This difficulty has catalyzed a significant escalation in research endeavors dedicated to engineering novel AQ-based compounds with augmented anticancer potency (11).
AQs offer several potential advantages over conventional chemotherapeutics in mitigating drug resistance. Unlike single-target agents, AQs frequently exhibit pleiotropic effects, impacting multiple cellular pathways. They include inducing apoptosis, arresting cell cycle progression, and triggering alternative cell death mechanisms such as paraptosis and autophagy (11,12). This multifaceted approach simultaneously disrupts multiple cellular processes, making it more challenging for cancer cells to develop resistance mechanisms that target a single pathway. Furthermore, AQs can interact with diverse cellular targets, including kinases and topoisomerases, further enhancing their anticancer activity and reducing the likelihood of resistance (11,13). Notably, some AQs demonstrate selective cytotoxicity toward cancer cells, minimizing off-target effects and potentially improving their safety profiles compared to traditional chemotherapeutics (14). These unique characteristics position AQs as promising candidates for developing novel cancer therapies that effectively address the challenge of drug resistance.
According to previous research, specific AQs represented anticancer properties by inhibiting cancer-associated proteins, including MMP2 (15), MMP13 (16), MAPK3 (17), and carbonic anhydrase (18). These proteins play crucial roles in cancer initiation, progression, and metastasis. This study extended previous investigations to examine the inhibitory effects of these compounds on Akt1, which is another key protein in cancer progression.
The pivotal role of Akt1 in cancer progression, coupled with the documented therapeutic efficacy of AQs in cancer treatment, presents a compelling rationale to investigate AQs as potential Akt1 inhibitors. This study harnesses the power of molecular docking analysis to evaluate the binding affinity of several prevalent AQs to the Akt1 ATP binding site. The results have been compared with those of standard drugs for Akt1 inhibition, thereby identifying promising lead compounds for further development into novel Akt1-targeted therapeutics for cancer treatment. These findings may identify promising herbal compounds that effectively target multiple cancer-associated markers.
Materials and Methods
Structural Preparations
The crystallographic structure of Akt1 was obtained from the Protein Data Bank (PDB ID: 4GV1, resolution: 1.49 Å) according to previous research (19). This structure, comprising a single 340-residue polypeptide chain, underwent energy optimization using Swiss-PdbViewer, version 4.1.0 (20). Next, the Discovery Studio Visualizer (21) was utilized to examine the interactions between the internal inhibitor, capivasertib, and the active site residues within the 4GV1 structure. This examination identified Leu156, Gly157, Gly162, Val164, Ala177, Met227, Ala230, Glu234, Glu278, Asn279, and Met281 as essential for ligand binding.
A set of 21 AQ compounds was curated for docking analyses to assess their binding affinities toward the Akt1 ATP-binding pocket. Molecular dockings were conducted and benchmarked against the established Akt1 inhibitors, resveratrol and capivasertib, which served as reference compounds. All small molecules were subjected to energy minimization following standard procedures using HyperChem software (22). Polar hydrogens were incorporated within the AutoDock 4.0 (16) environment, and Kollman charges were assigned to them. The structure underwent further processing to enable rotational freedom for the small molecules and account for their localized charges. Eventually, the Protein Data Bank Quick Preparation Tool was employed to generate input files for docking analyses, executed through the Cygwin64 Terminal interface.
Molecular Docking and Interaction Mode Analyses
AutoDock 4.0 was utilized on a high-end Windows workstation equipped with an Intel Core i7 CPU to run molecular docking simulations and assess the binding affinities of 21 AQ compounds, capivasertib, and resveratrol to the Akt1 catalytic cleft. A 60 Å x 60 Å x 60 Å cubic grid box was placed at coordinates (20.556 Å, 5.617 Å, 11.523 Å) with a grid spacing of 0.375 Å to allow for different possible ligand orientations inside the active site.
To address ligand flexibility, 50 different conformations were produced for every molecule. The binding free energy (ΔGbinding) for each molecule was calculated, which measures the potency of ligand-protein interactions. The conformation belonging to the most populated cluster and having the highest negative ΔGbinding value from each set of conformations was chosen for further examination.
The interaction patterns between the selected ligand conformations and the Akt1 protein were meticulously rendered and analyzed using the Discovery Studio Visualizer. This methodology illustrated potential binding modes in three dimensions, offering valuable insights into the molecular interactions that govern ligand recognition and affinity.
Results
Binding Affinity Assessments
It was found that 11 AQs had a substantial binding affinity to the enzyme’s catalytic site and could inhibit Akt1 activity at nanomolar concentrations. These chemicals were identified as hypericin, sennidin B, rhodoptilometrin, Aloe-emodin 8-glucoside, alizarin, chrysophanol, rhein, purpurin, and emodic acid. Of all the examined AQs, sennidin B and hypericin had the most potent inhibitory effects against the Akt1 catalytic cleft, with ΔGbinding values of -10.36 kcal/mol and -11.19 kcal/mol, respectively.
The reference drugs, capivasertib and resveratrol, exhibited inhibition constants of 26.16 nM and 18.49 µM, respectively. Additionally, their binding free energy values were measured at 10.34 kcal/mol and -6.46 kcal/mol for capivasertib and resveratrol, respectively.
All the AQs examined in this investigation, except for damnacanthal and emodin-8-glucoside, had a higher binding affinity to the enzyme’s active site than the control inhibitor (Table 1). These results imply that some of the intended AQs might be regarded as putative Akt1 inhibitors, and more investigation into their potential therapeutic uses might be necessary. Figure 1 compares the most remarkable binding affinities found for the identified AQs with the ΔGbinding values of the reference compounds.
Table 1.
Information on the Energies and Ki Values Between the Akt1 Catalytic Cleft, Resveratrol, and the Studied Anthraquinones
|
PubChem ID
|
Ligand Name
|
ΔGbinding
|
Ki Value
|
Intermolecular Energy (kcal/mol)
|
Total Internal Energy (kcal/mol)
|
Torsional Free Energy (kcal/mol)
|
Unbound System's Energy (kcal/mol)
|
| 3663 |
Hypericin |
-11.19 |
6.30 nM |
-9.51 |
-4.32 |
2.39 |
-0.26 |
| 10459879 |
Sennidin B |
-10.36 |
25.62 nM |
-12.69 |
-3.99 |
3.88 |
-2.45 |
| 126456371 |
Aloe emodin 8-glucoside |
-9.9 |
55.55 nM |
-8.93 |
-5.93 |
3.58 |
-1.38 |
| 442731 |
Pulmatin (Chrysophanol-8-0-glucoside) |
-9.84 |
61.46 nM |
-8.61 |
-5.71 |
2.98 |
-1.5 |
| 101286218 |
Rhodoptilometrin |
-9.4 |
128.36 nM |
-10.2 |
-2.07 |
2.39 |
-0.49 |
| 10207 |
Aloe-emodin |
-8.97 |
265.91 nM |
-9.1 |
-1.99 |
1.79 |
-0.33 |
| 6293 |
Alizarin |
-8.91 |
295.49 nM |
-8.26 |
-2.11 |
1.19 |
-0.28 |
| 10208 |
Chrysophanol |
-8.73 |
396.91 nM |
-8.29 |
-2.01 |
1.19 |
-0.37 |
| 10168 |
Rhein |
-8.46 |
632.09 nM |
-8.51 |
-2.11 |
2.09 |
-0.07 |
| 6683 |
Purpurin |
-8.38 |
722.45 nM |
-7.67 |
-2.5 |
1.49 |
-0.29 |
| 361510 |
Emodic acid |
-8.19 |
985.76 nM |
-8.41 |
-2.23 |
2.39 |
-0.06 |
| 3220 |
Emodin |
-8.1 |
1.15 uM |
-7.36 |
-2.44 |
1.49 |
-0.2 |
| 2950 |
Danthron |
-8.09 |
1.17 uM |
-7.8 |
-1.98 |
1.19 |
-0.5 |
| 10639 |
Physcion |
-8 |
1.36 uM |
-8.37 |
-1.42 |
1.49 |
-0.29 |
| 3083575 |
Obtusifolin |
-7.88 |
1.66 uM |
-7.69 |
-2.24 |
1.49 |
-0.56 |
| 92826 |
Sennidin A |
-7.27 |
4.65 uM |
-10.73 |
-5.2 |
3.88 |
-4.78 |
| 124062 |
Rubiadin |
-7.22 |
5.10 uM |
-8.6 |
-1.11 |
1.19 |
-1.3 |
| 160712 |
Nordamnacanthal |
-7.01 |
7.29 uM |
-.61 |
-2.15 |
1.79 |
-1.96 |
| 442753 |
Knipholone |
-6.74 |
11.55 uM |
-9.6 |
-5.02 |
2.98 |
-4.9 |
| 2948 |
Damnacanthal |
-6.37 |
21.24 uM |
-8.29 |
-1.33 |
1.79 |
-1.46 |
| 99649 |
Emodin-8-glucoside |
-1.9 |
40.77 mM |
-0.1 |
-6.56 |
3.28 |
-1.48 |
| 25227436 |
Capivasertib (Ctrl + ) |
-10.34 |
26.16 nM |
-10.40 |
-2.95 |
+ 2.39 |
-0.62 |
| 445154 |
Resveratrol (Ctrl + ) |
-6.46 |
18.49 uM |
-7.99 |
-0.47 |
1.79 |
-0.22 |
Note. Akt1: RAC-alpha serine/threonine-protein kinase; Ki: Inhibition constant; Ctrl: Control.
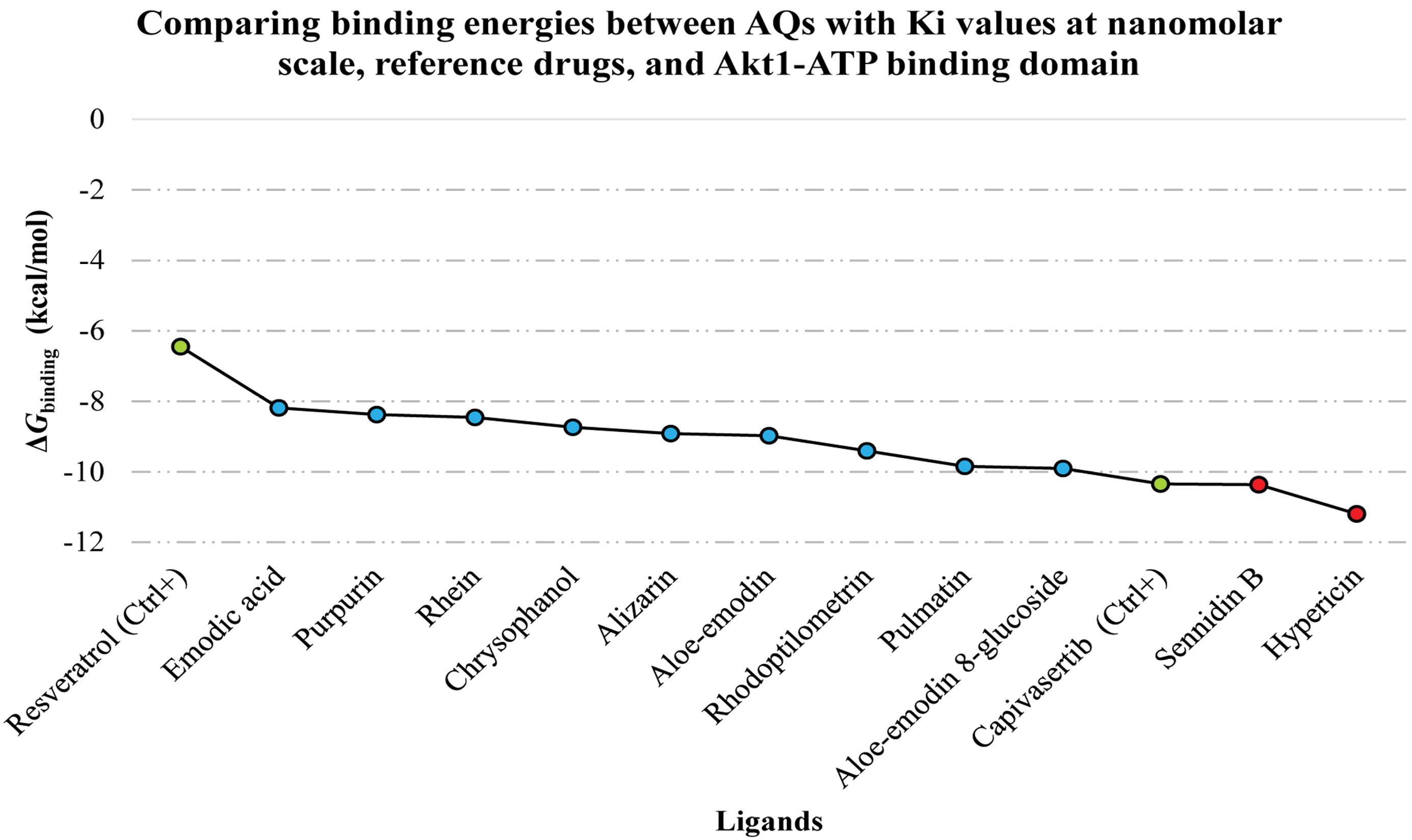
Figure 1.
The Top-Ranked AQs Evaluated for Their Binding Affinity to the Akt1 Catalytic Cleft Compared to Reference Inhibitors. Note. AQ: Anthraquinone; Ki: Inhibition constant; Akt1: RAC-alpha serine/threonine-protein kinase; Ctrl: Control
.
The Top-Ranked AQs Evaluated for Their Binding Affinity to the Akt1 Catalytic Cleft Compared to Reference Inhibitors. Note. AQ: Anthraquinone; Ki: Inhibition constant; Akt1: RAC-alpha serine/threonine-protein kinase; Ctrl: Control
Interaction Modes
A comparative analysis was conducted regarding the interactions between the top-ranked AQs, control inhibitors, and critical residues within the Akt1 catalytic cleft. It was observed that neither hypericin nor sennidin B formed any electrostatic interactions with the enzyme’s active site. Hypericin exhibited three hydrogen bonds and two hydrophobic interactions with residues inside the Akt1 catalytic cleft. In contrast, sennidin B formed six hydrogen bonds and one hydrophobic interaction with amino acids within the enzyme’s active site. The control inhibitor, resveratrol, displayed a more diverse interaction profile, demonstrating two hydrogen bonds, five hydrophobic interactions, and two electrostatic interactions with amino acids within the Akt1 catalytic cleft. Capivasertib also formed five H-bonds and 11 hydrophobic interactions with the target protein (Table 2).
Table 2.
Top-Ranked Anthraquinones, a Reference Medication, and Akt1 Catalytic Cleft Interactions
|
Ligand Name
|
Hydrogen Bond (Distance in Angstrom)
|
Hydrophobic Interaction (Distance in Angstrom)
|
Electrostatic (Distance in Angstrom)
|
| Hypericin |
Asp274 (4.71), Asp292 (2.97), and Gly162 (2.94) |
Phe161 (5.13) and Leu295 (6.04) |
Na |
| Sennidin B |
Thr160 (2.84), Glu278 (2.87), Glu234 (4.41), Gly157 (4.25), Val164 (3.58), and Gly162 (3.21) |
Val164 (5.74) |
Na |
| Capivasertib (Ctrl + ) |
Glu228 (4.73), Ala230 (4.04), Glu234 (4.68), Glu278 (4.49), and Tyr229 (4.15) |
Leu181 (5.07), Lys179 (4.75, 5.98), Val164 (4.71, 6.07), Met281 (6.66, 5.68, 6.11), Ala177 (4.71, 6.1), and Ala230 (5.32); |
Na |
| Resveratrol (Ctrl + ) |
Ala230 (3.49) and Asn279 (4.13) |
Met281 (6.83), Val164 (4.35), Leu156 (4.96), Ala177 (6.08), and Ala230 (6.45) |
Glu234 (6.33) and Asp292 (6.58) |
Note. Akt1: RAC-alpha serine/threonine-protein kinase; Na: Not available; Ctrl: Control.
For clarity, Figure 2 displays two- and three-dimensional representations of the compounds hypericin, sennidin B, and reference inhibitors inside the receptor’s active site. These representations show the spatial arrangement of these chemicals within the enzyme’s binding pocket.
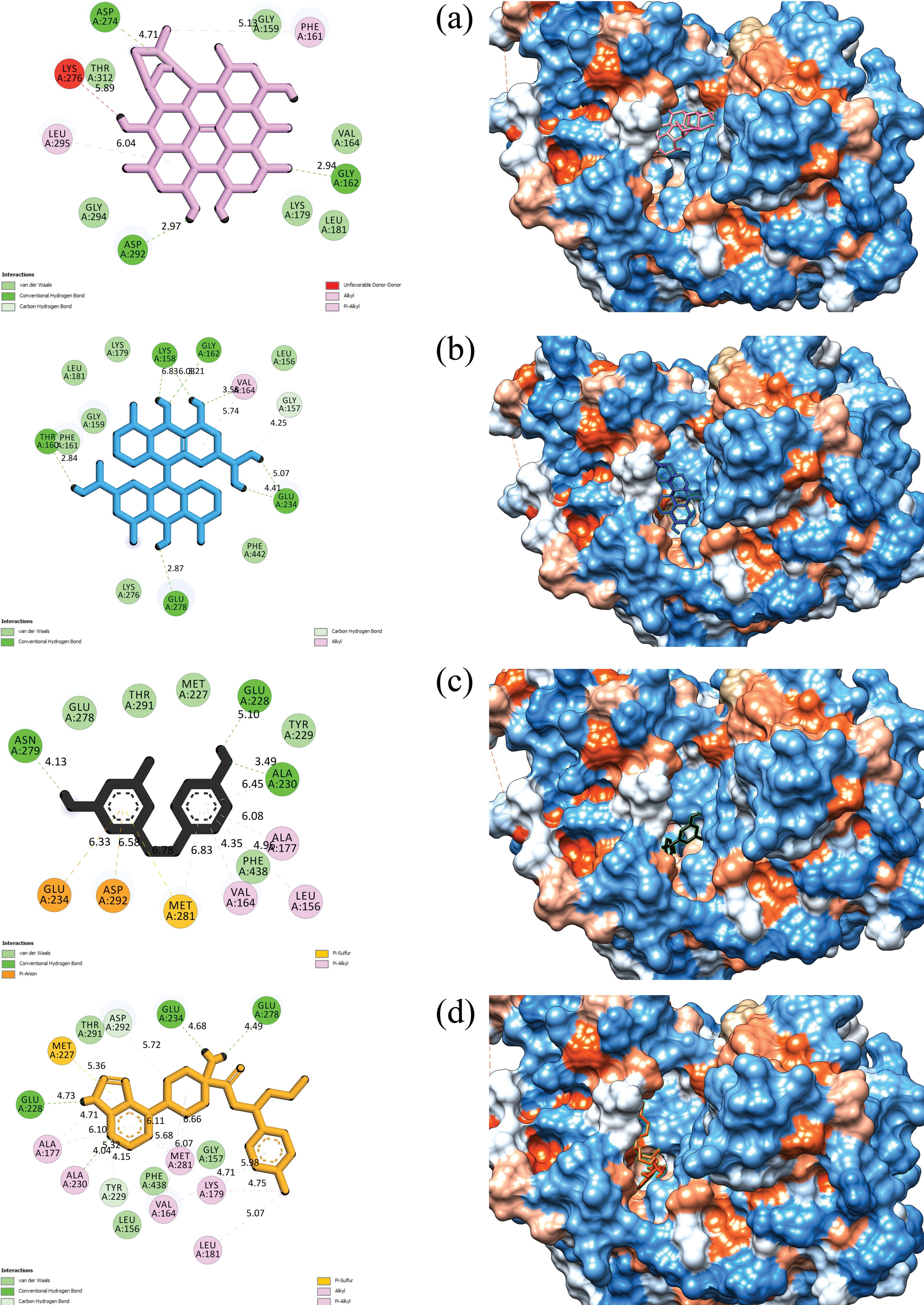
Figure 2.
Two- and three-Dimensional Views of (a) Hypericin, (b) Sennidin B, (c) Resveratrol, and (d) Capivasertib Inside the Akt1 Active Site. Note. Akt1: RAC-alpha serine/threonine-protein kinase
.
Two- and three-Dimensional Views of (a) Hypericin, (b) Sennidin B, (c) Resveratrol, and (d) Capivasertib Inside the Akt1 Active Site. Note. Akt1: RAC-alpha serine/threonine-protein kinase
Discussion
Akt1 inhibition in cancer treatment has emerged as a promising approach due to its critical function within the PI3K/Akt/mTOR pathway, often deregulated in many cancers. As a serine/threonine kinase, Akt1 regulates cell survival, proliferation, and metabolic processes (23); thus, targeting Akt1 has shown therapeutic potential in preclinical studies (24,25). This research focused on computational methods to evaluate the inhibitory potential of various AQs targeting the Akt1 catalytic site. The findings were compared with those related to resveratrol, a well-established Akt inhibitor, to identify promising lead compounds for further exploration.
Based on the results, hypericin and sennidin B displayed extreme binding affinity to the Akt1 catalytic cleft, with ΔGbinding values of -11.19 kcal/mol and -10.36 kcal/mol, respectively. These were followed by Aloe-emodin 8-glucoside (ΔGbinding = -9.9 kcal/mol), Chrysophanol-8-0-glucoside (ΔGbinding = -9.84 kcal/mol), and Rhodoptilometrin (ΔGbinding = -9.4 kcal/mol), which also emerged as promising Akt1 inhibitors. Capivasertib and resveratrol represented ΔGbinding values of -10.34 kcal/mol and -6.46 kcal/mol.
AQs have significantly affected Akt1 signaling, crucial in cancer cell survival and proliferation. In this regard, the results of a study by de Souza Alves et al (26) demonstrated that AQ derivatives can effectively reduce Akt phosphorylation, inhibiting the PI3K/Akt pathway in various cellular contexts, including allergic airway disease models. Hypericin, a well-studied AQ, has been reported to inhibit Akt1 activity in cancer cells, leading to increased apoptosis through the downregulation of anti-apoptotic proteins such as Bcl-2 and the activation of pro-apoptotic pathways (27,28). Additionally, another AQ derivative, SZ-685C, was found to induce apoptosis by modulating Akt signaling, effectively overcoming drug resistance in cancer cells (29). These findings suggest that AQs not only target Akt1 directly but also influence downstream signaling pathways, positioning them as promising candidates for further exploration in cancer therapy, aiming at modulating the Akt1 pathway and potentially reducing drug resistance.
Hypericum perforatum, a plant native to Europe, is a promising source of hypericin, a compound with multifaceted anticancer activity (30). Traditionally used for medicinal purposes, hypericin’s effects include direct cell death induced by reactive oxygen species upon light activation and the modulation of cell death pathways (31,32). A significant clinical trial by Kim et al (33) investigated a topical hypericin ointment activated by visible light for treating early-stage cutaneous T-cell lymphoma. This phase III randomized study involving 169 patients confirmed a substantial improvement in lesion response rates compared to a placebo group. After six weeks, the index lesion response rate was 16% for the hypericin group compared to only 4% for the placebo group (P = 0.04). Further treatment cycles significantly increased response rates to 40% and 49%, respectively (P < 0.001). Supporting preclinical data has been reviewed by Wu et al (34). Accordingly, hypericin triggers apoptosis through caspase-3 and caspase-4 activation, additionally disrupting mitochondrial function to induce cancer cell death. Furthermore, Olek et al (35) explored the immune system-modulating effects of hypericin-based photodynamic therapy on OSCCs (35). Their findings revealed significant changes in cytokine secretion profiles following treatment. According to the present results, hypericin’s strong binding affinity appears to be attributed to its formation of three hydrogen bonds with vital amino acids, namely, Gly162, Asp274, and Asp292, in the Akt1 catalytic cleft. Additionally, it exhibited two hydrophobic interactions with Phe161 and Leu295, further stabilizing the complex.
Cassia senna species, particularly Cassia acutifolia and Cassia angustifolia, are widely cultivated sources of Sennoside B, found in regions such as Somalia, the Arabian Peninsula, South India, and Pakistan (36). Chen et al (37) investigated the effects of Sennoside B on cell signaling using human osteosarcoma MG63 cells. The researchers discovered that Sennoside B could significantly inhibit the platelet-derived growth factor (PDGF)-BB-induced activation (phosphorylation) of the PDGF receptor. This inhibition cascaded down to downstream molecules such as AKT, STAT-5, and ERK1/2, ultimately leading to a substantial reduction in cell proliferation. These findings indicate that Sennoside B might disrupt crucial signaling pathways involved in cancer cell growth and division, making it a potential therapeutic candidate for conditions driven by PDGF signaling. It is noteworthy that Sennoside B is a dimeric glycoside consisting of two rhein molecules linked by a dianthrone structure. In the colon, bacterial hydrolysis converts Sennoside B to rhein anthrone, the active metabolite that is responsible for its laxative effect (38). Sennidin B, an aglycone derivative formed during Sennoside B’s breakdown, is an intermediate product in this pathway (39). Interestingly, sennidin B’s binding appears to be stabilized by six H-bonds and one hydrophobic interaction with critical residues, including Gly157, Thr160, Gly162, Val164, Glu234, and Glu278, in the Akt1 catalytic cleft (39).
Reynoutria japonica Houtt. (Polygonaceae) and polygoni multiflori radix are recognized as essential sources of Aloe-emodin 8-glucoside (40,41). Kim et al (42) demonstrated that Aloe-emodin 3-O-glucoside significantly inhibited cell growth and migration and induced apoptosis in non-small-cell lung cancer models, likely by suppressing the MEK/ERK and Akt signaling pathways. Given the structural similarities between these compounds, Aloe-emodin 8-O-glucoside might possess analogous mechanisms of action.
The reference drug resveratrol formed a network of interactions with the Akt1 catalytic cleft. This includes two hydrogen bonds with Ala230 and Asn279, potentially contributing to its binding affinity. Additionally, resveratrol exhibited five hydrophobic interactions with Leu156, Val164, Met281, Ala177, and Ala230, further stabilizing the complex. Interestingly, it also displayed two electrostatic interactions with Glu234 and Asp292, suggesting a more intricate binding mode than hypericin and sennidin B.
While previous studies have confirmed the anticancer activity of certain AQs, this study has provided several key advancements. Firstly, a comprehensive in silico analysis of a diverse panel of 21 AQs was conducted, systematically evaluating their binding affinity and interaction modes with Akt1. This approach allowed for identifying novel potential Akt1 inhibitors within this class of compounds that had not been previously reported to target Akt1. Secondly, this study provided detailed molecular insights into the binding interactions of these AQs with Akt1, revealing crucial hydrogen bonding and hydrophobic interactions that contribute to their inhibitory potential. This information can guide the design and optimization of novel Akt1 inhibitors with improved potency and specificity. Finally, by comparing the binding affinities of these AQs with established Akt1 inhibitors, this study presented a valuable framework for prioritizing promising candidates for further experimental validation.
Conclusion
This study provided compelling evidence for the potential of AQs as novel Akt1 inhibitors in cancer treatment. Through molecular docking analysis, this study identified several AQs with superior binding affinities to the Akt1 catalytic cleft compared to the established inhibitors, capivasertib and resveratrol. The diverse interaction profiles observed among the AQs suggest multiple mechanisms by which these compounds may inhibit Akt1 activity. This diversity could potentially address challenges associated with drug resistance in cancer therapy. Moreover, the natural origin of these compounds may offer advantages in terms of reduced side effects and improved tolerability compared to synthetic alternatives.
Acknowledgments
The authors acknowledge the support of the Deputy of Research and Technology, Research Center for Molecular Medicine, and Dental Research Center, Hamadan University of Medical Sciences, Hamadan, Iran.
Authors’ Contribution
Conceptualization: Amir Taherkhani, Shokoofeh Jamshidi.
Data curation: Amir Taherkhani, Fatemeh Mahfouzi.
Formal analysis: Amir Taherkhani, Fatemeh Mahfouzi.
Investigation: Amir Taherkhani, Shokoofeh Jamshidi.
Methodology: Amir Taherkhani, Fatemeh Mahfouzi.
Project administration: Amir Taherkhani, Shokoofeh Jamshidi.
Resources: Amir Taherkhani.
Software: Amir Taherkhani, Fatemeh Mahfouzi.
Supervision: Amir Taherkhani, Shokoofeh Jamshidi.
Validation: Amir Taherkhani, Shokoofeh Jamshidi, Setareh Shojaei.
Visualization: Amir Taherkhani, Fatemeh Mahfouzi.
Writing–original draft: Amir Taherkhani.
Writing–review & editing: Shokoofeh Jamshidi, Setareh Shojaei.
Competing Interests
The authors declare that they have no competing interests.
Data Availability Statement
The datasets used and/or analyzed during the currentstudy are available from the corresponding author upon reasonable request.
Ethical Approval
This study was confirmed by the Ethics Committee of Hamadan University of Medical Sciences, Hamadan, Iran (IR.UMSHA.REC.1402.232).
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
- Nowak-Perlak M, Ziółkowski P, Woźniak M. A promising natural anthraquinones mediated by photodynamic therapy for anti-cancer therapy. Phytomedicine 2023; 119:155035. doi: 10.1016/j.phymed.2023.155035 [Crossref] [ Google Scholar]
- Massari F, Ciccarese C, Santoni M, Iacovelli R, Mazzucchelli R, Piva F. Metabolic phenotype of bladder cancer. Cancer Treat Rev 2016; 45:46-57. doi: 10.1016/j.ctrv.2016.03.005 [Crossref] [ Google Scholar]
- Kim DD, Eng C. The promise of mTOR inhibitors in the treatment of colorectal cancer. Expert Opin Investig Drugs 2012; 21(12):1775-88. doi: 10.1517/13543784.2012.721353 [Crossref] [ Google Scholar]
- Singh SK, Apata T, Singh S, McFadden M, Singh R. Clinical implication of metformin in relation to diabetes mellitus and ovarian cancer. Biomedicines 2021; 9(8):1020. doi: 10.3390/biomedicines9081020 [Crossref] [ Google Scholar]
- Dey S, Singh AK, Singh AK, Rawat K, Banerjee J, Agnihotri V. Critical pathways of oral squamous cell carcinoma: molecular biomarker and therapeutic intervention. Med Oncol 2022; 39(3):30. doi: 10.1007/s12032-021-01633-4 [Crossref] [ Google Scholar]
- Su YC, Lee WC, Wang CC, Yeh SA, Chen WH, Chen PJ. Targeting PI3K/AKT/mTOR signaling pathway as a radiosensitization in head and neck squamous cell carcinomas. Int J Mol Sci 2022; 23(24):15749. doi: 10.3390/ijms232415749 [Crossref] [ Google Scholar]
- Tian X, Wang R, Gu T, Ma F, Laster KV, Li X. Costunolide is a dual inhibitor of MEK1 and AKT1/2 that overcomes osimertinib resistance in lung cancer. Mol Cancer 2022; 21(1):193. doi: 10.1186/s12943-022-01662-1 [Crossref] [ Google Scholar]
- Smyth LM, Tamura K, Oliveira M, Ciruelos EM, Mayer IA, Sablin MP. Capivasertib, an AKT kinase inhibitor, as monotherapy or in combination with fulvestrant in patients with AKT1 (E17K)-mutant, ER-positive metastatic breast cancer. Clin Cancer Res 2020; 26(15):3947-57. doi: 10.1158/1078-0432.Ccr-19-3953 [Crossref] [ Google Scholar]
- Nowak-Perlak M, Ziółkowski P, Woźniak M. A promising natural anthraquinones mediated by photodynamic therapy for anti-cancer therapy. Phytomedicine 2023; 119:155035. doi: 10.1016/j.phymed.2023.155035 [Crossref] [ Google Scholar]
- Wang P, Wei J, Hua X, Dong G, Dziedzic K, Wahab AT. Plant anthraquinones: classification, distribution, biosynthesis, and regulation. J Cell Physiol 2024; 239(10):e31063. doi: 10.1002/jcp.31063 [Crossref] [ Google Scholar]
- Malik MS, Alsantali RI, Jassas RS, Alsimaree AA, Syed R, Alsharif MA. Journey of anthraquinones as anticancer agents - a systematic review of recent literature. RSC Adv 2021; 11(57):35806-27. doi: 10.1039/d1ra05686g [Crossref] [ Google Scholar]
- Tian W, Wang C, Li D, Hou H. Novel anthraquinone compounds as anticancer agents and their potential mechanism. Future Med Chem 2020; 12(7):627-44. doi: 10.4155/fmc-2019-0322 [Crossref] [ Google Scholar]
- Castro DT, Leite DF, da Silva Baldivia D, Dos Santos HF, Balogun SO, da Silva DB. Structural characterization and anticancer activity of a new anthraquinone from Senna velutina (Fabaceae). Pharmaceuticals (Basel) 2023; 16(7):951. doi: 10.3390/ph16070951 [Crossref] [ Google Scholar]
- Huang Q, Lu G, Shen HM, Chung MC, Ong CN. Anti-cancer properties of anthraquinones from rhubarb. Med Res Rev 2007; 27(5):609-30. doi: 10.1002/med.20094 [Crossref] [ Google Scholar]
- Jamshidi S, Rostami A, Shojaei S, Taherkhani A, Taherkhani H. Exploring natural anthraquinones as potential MMP2 inhibitors: a computational study. Biosystems 2024; 235:105103. doi: 10.1016/j.biosystems.2023.105103 [Crossref] [ Google Scholar]
- Taherkhani A, Moradkhani S, Orangi A, Jalalvand A. In silico study of some natural anthraquinones on matrix metalloproteinase inhibition. Res J Pharmacogn 2021; 8(4):37-51. doi: 10.22127/rjp.2021.288366.1705 [Crossref] [ Google Scholar]
- Vaziri-Amjad S, Moradi-Najmi M, Taherkhani A. Natural anthraquinones as promising MAPK3 inhibitors for complementary cancer therapy. J Chem 2023; 2023(1):6683470. doi: 10.1155/2023/6683470 [Crossref] [ Google Scholar]
- Yarmohammadi E, Khanjani M, Khamverdi Z, Savari M, Taherkhani A. Herbal metabolites as potential carbonic anhydrase inhibitors: promising compounds for cancer and metabolic disorders. J Obes Metab Syndr 2023; 32(3):247-58. doi: 10.7570/jomes23029 [Crossref] [ Google Scholar]
- Addie M, Ballard P, Buttar D, Crafter C, Currie G, Davies BR. Discovery of 4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (AZD5363), an orally bioavailable, potent inhibitor of Akt kinases. J Med Chem 2013; 56(5):2059-73. doi: 10.1021/jm301762v [Crossref] [ Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997; 18(15):2714-23. doi: 10.1002/elps.1150181505 [Crossref] [ Google Scholar]
- Studio D. Discovery studio. Accelrys [21] 2008;420.
- Laxmi D, Priyadarshy S. HyperChem 603. Biotech Software & Internet Report 2002; 3(1):5-9. doi: 10.1089/152791602317250351 [Crossref] [ Google Scholar]
- Shariati M, Meric-Bernstam F. Targeting AKT for cancer therapy. Expert Opin Investig Drugs 2019; 28(11):977-88. doi: 10.1080/13543784.2019.1676726 [Crossref] [ Google Scholar]
- Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther 2012; 11(4):873-87. doi: 10.1158/1535-7163.Mct-11-0824-t [Crossref] [ Google Scholar]
- Tang F, Wang Y, Hemmings BA, Rüegg C, Xue G. PKB/Akt-dependent regulation of inflammation in cancer. Semin Cancer Biol 2018; 48:62-9. doi: 10.1016/j.semcancer.2017.04.018 [Crossref] [ Google Scholar]
- de Souza Alves CC, Collison A, Hatchwell L, Plank M, Morten M, Foster PS. Inhibiting AKT phosphorylation employing non-cytotoxic anthraquinones ameliorates TH2 mediated allergic airways disease and rhinovirus exacerbation. PLoS One 2013; 8(11):e79565. doi: 10.1371/journal.pone.0079565 [Crossref] [ Google Scholar]
- Buľková V, Vargová J, Babinčák M, Jendželovský R, Zdráhal Z, Roudnický P. New findings on the action of hypericin in hypoxic cancer cells with a focus on the modulation of side population cells. Biomed Pharmacother 2023; 163:114829. doi: 10.1016/j.biopha.2023.114829 [Crossref] [ Google Scholar]
- Merhi F, Tang R, Piedfer M, Mathieu J, Bombarda I, Zaher M. Hyperforin inhibits Akt1 kinase activity and promotes caspase-mediated apoptosis involving Bad and Noxa activation in human myeloid tumor cells. PLoS One 2011; 6(10):e25963. doi: 10.1371/journal.pone.0025963 [Crossref] [ Google Scholar]
- Zhu X, He Z, Wu J, Yuan J, Wen W, Hu Y. A marine anthraquinone SZ-685C overrides adriamycin-resistance in breast cancer cells through suppressing Akt signaling. Mar Drugs 2012; 10(4):694-711. doi: 10.3390/md10040694 [Crossref] [ Google Scholar]
- Barnes J, Anderson LA, Phillipson JD. St John’s wort (Hypericum perforatum L): a review of its chemistry, pharmacology and clinical properties. J Pharm Pharmacol 2001; 53(5):583-600. doi: 10.1211/0022357011775910 [Crossref] [ Google Scholar]
- Wölfle U, Seelinger G, Schempp CM. Topical application of St John’s wort (Hypericum perforatum). Planta Med 2014; 80(2-3):109-20. doi: 10.1055/s-0033-1351019 [Crossref] [ Google Scholar]
- Dong X, Zeng Y, Zhang Z, Fu J, You L, He Y. Hypericin-mediated photodynamic therapy for the treatment of cancer: a review. J Pharm Pharmacol 2021; 73(4):425-36. doi: 10.1093/jpp/rgaa018 [Crossref] [ Google Scholar]
- Kim EJ, Mangold AR, DeSimone JA, Wong HK, Seminario-Vidal L, Guitart J. Efficacy and safety of topical hypericin photodynamic therapy for early-stage cutaneous T-cell lymphoma (Mycosis fungoides): the FLASH phase 3 randomized clinical trial. JAMA Dermatol 2022; 158(9):1031-9. doi: 10.1001/jamadermatol.2022.2749 [Crossref] [ Google Scholar]
- Wu JJ, Zhang J, Xia CY, Ding K, Li XX, Pan XG. Hypericin: a natural anthraquinone as promising therapeutic agent. Phytomedicine 2023; 111:154654. doi: 10.1016/j.phymed.2023.154654 [Crossref] [ Google Scholar]
- Olek M, Machorowska-Pieniążek A, Czuba ZP, Cieślar G, Kawczyk-Krupka A. Immunomodulatory effect of hypericin-mediated photodynamic therapy on oral cancer cells. Pharmaceutics 2023; 16(1):42. doi: 10.3390/pharmaceutics16010042 [Crossref] [ Google Scholar]
- Franz G. The Senna drug and its chemistry. Pharmacology 1993; 47 Suppl 1:2-6. doi: 10.1159/000139654 [Crossref] [ Google Scholar]
- Chen YC, Chang CN, Hsu HC, Chiou SJ, Lee LT, Hseu TH. Sennoside B inhibits PDGF receptor signaling and cell proliferation induced by PDGF-BB in human osteosarcoma cells. Life Sci 2009; 84(25-26):915-22. doi: 10.1016/j.lfs.2009.04.003 [Crossref] [ Google Scholar]
- Diao F, Xie Q, Wu X, Liu C, Li X, Li Q. [Determination of sennoside B, sennoside A and physcion in slimming health foods]. Wei Sheng Yan Jiu 2020; 49(5):809-14. doi: 10.19813/j.cnki.weishengyanjiu.2020.05.020 [Crossref] [ Google Scholar]
- Alam P, Noman OM, Herqash RN, Almarfadi OM, Akhtar A, Alqahtani AS. Response surface methodology (RSM)-based optimization of ultrasound-assisted extraction of sennoside A, sennoside B, aloe-emodin, emodin, and chrysophanol from Senna alexandrina (aerial parts): HPLC-UV and antioxidant analysis. Molecules 2022; 27(1):298. doi: 10.3390/molecules27010298 [Crossref] [ Google Scholar]
- Okon E, Koval M, Wawruszak A, Slawinska-Brych A, Smolinska K, Shevera M. Emodin-8-O-glucoside-isolation and the screening of the anticancer potential against the nervous system tumors. Molecules 2023; 28(21):7366. doi: 10.3390/molecules28217366 [Crossref] [ Google Scholar]
- Wang X, Zhao G, Ju C, Dong L, Liu Y, Ding Z. Reduction of emodin-8-O-ß-D-glucoside content participates in processing-based detoxification of polygoni multiflori radix. Phytomedicine 2023; 114:154750. doi: 10.1016/j.phymed.2023.154750 [Crossref] [ Google Scholar]
- Kim HJ, Choi JW, Ree J, Lim JS, Lee J, Kim JI. Aloe emodin 3-O-glucoside inhibits cell growth and migration and induces apoptosis of non-small-cell lung cancer cells via suppressing MEK/ERK and Akt signalling pathways. Life Sci 2022; 300:120495. doi: 10.1016/j.lfs.2022.120495 [Crossref] [ Google Scholar]